Multi-quantitative tracks
JBrowse can show "Multi-quantitative tracks" which is a single track composed of multiple quantitative signals, which have their Y-scalebar synchronized.
There are 5 rendering modes for the multi-quantitative tracks.
- xyplot
- multirowxyplot
- multiline
- multirowline
- multidensity
You can interactively change these settings through the track menu.
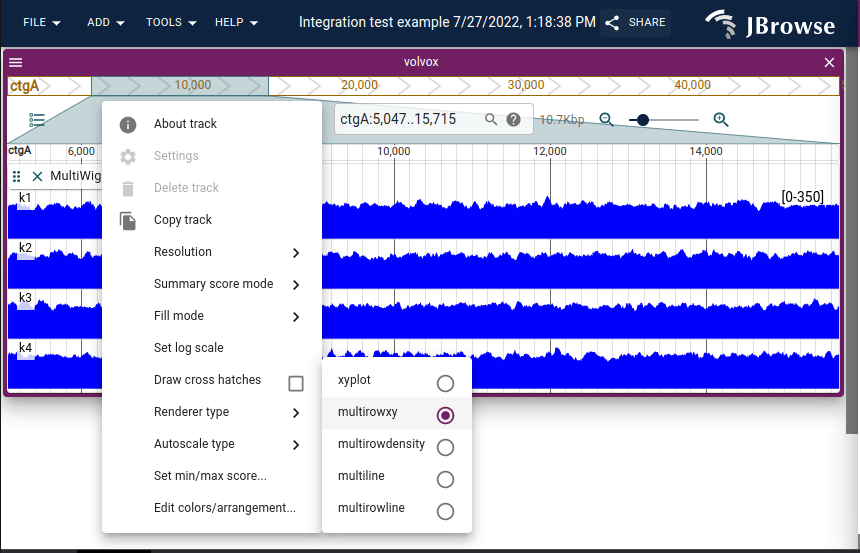
With the "multi-row" settings (multirowxyplot, multirowline, multidensity) the track colors are not modified. For the overlapping (xyplot, multiline), the tracks will be autoassigned a color from the palette. You can manually customize the subtrack colors from the track menu as well.
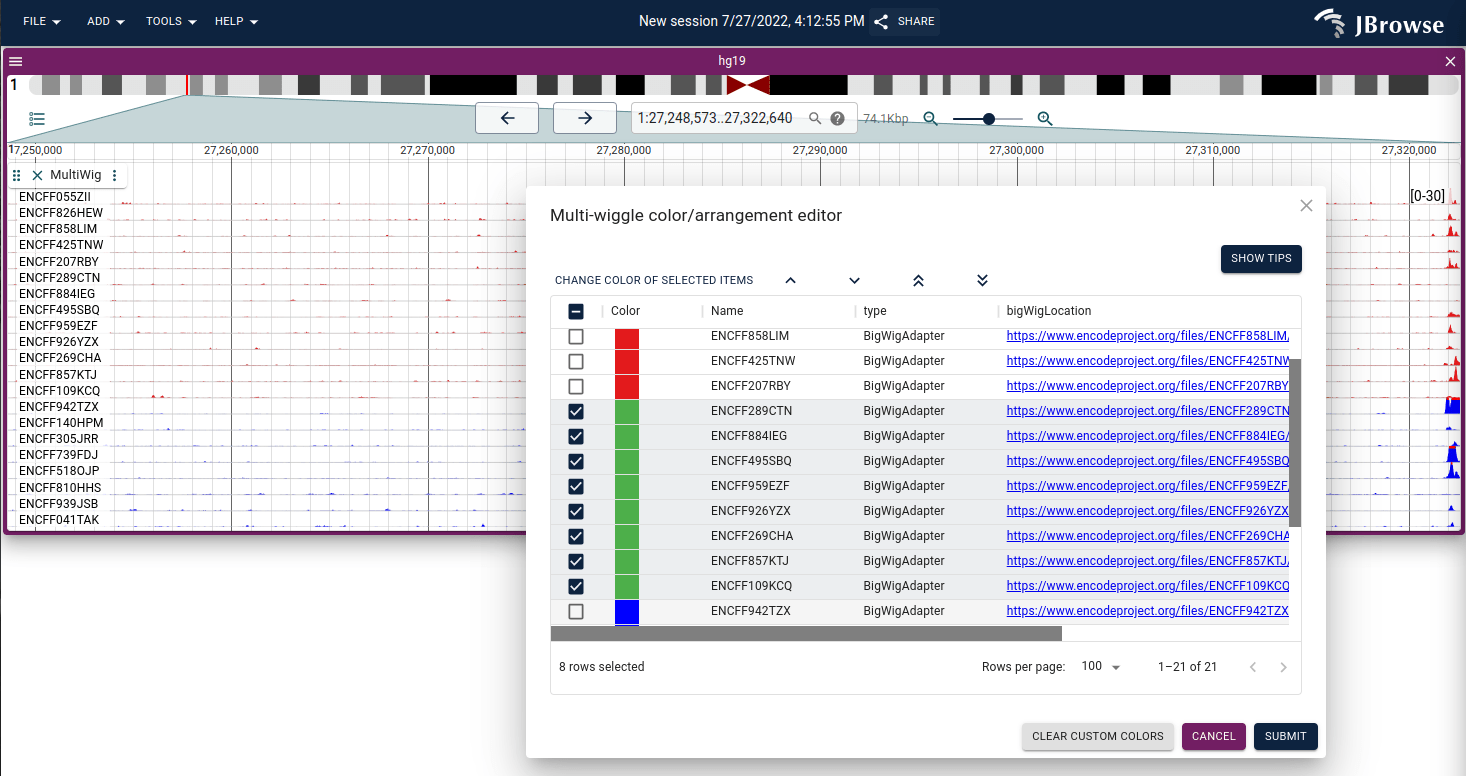
Oftentimes, one of the outliers on one of the subtracks may affect the Y-scalebar too much, so it is often helpful to use the "Autoscale type → Local +/- 3SD" setting (3 standard deviations are displayed). Manually configuring the min or max scores is available via the track menu also.
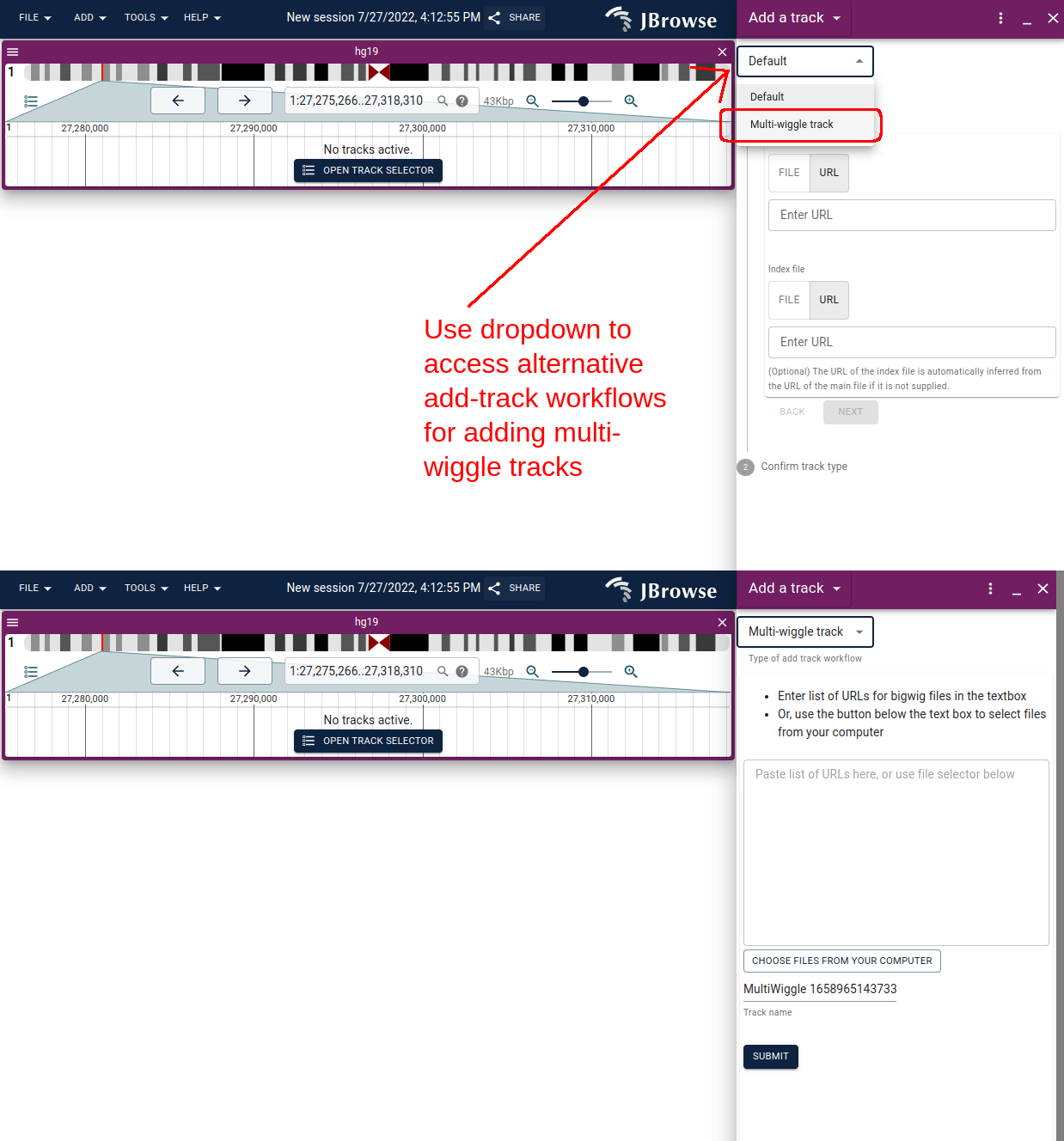
Adding multi-quantitative tracks via the UI
There are several ways to create multi-quantitative tracks from scratch.
- Using the add track panel to open up a list of URLs for bigwig files, or from several local tracks from your machine
- Using the track selector to add multiple tracks to your current selection, and then creating a multi-wiggle track from the tracks in your selection
- Hardcoding the multiwiggle track in your config file (see multi-quantitative track configuration for more info)
Loading bedMethyl as a multi-quantitative track
modkit pileup produces a
bedMethyl file — a
tab-separated BED format where each row reports the methylation fraction at a
single CpG position for one modification type (e.g. 5mC or 5hmC). Because the
format is a BED file it can be loaded with BedTabixAdapter, and because each
modification type produces its own score column the file maps naturally to a
MultiQuantitativeTrack.
Generating the file
modkit pileup sample.bam output.bed --ref reference.fa --preset traditional
bgzip output.bed
tabix -p bed output.bed.gz
The --preset traditional flag produces 5mC calls (5hmC is combined into the
5mC fraction). Omit it for separate 5mC and 5hmC rows.
Add-track UI
In the add-track dialog (File → Open track...), paste the URL to your
.bedmethyl.gz file. JBrowse detects the .bedmethyl.gz extension and
automatically selects BedTabixAdapter and MultiQuantitativeTrack — no
manual selection needed.
Config example
{
"type": "MultiQuantitativeTrack",
"trackId": "sample_modkit",
"name": "CpG methylation (modkit)",
"assemblyNames": ["hg38"],
"adapter": {
"type": "BedTabixAdapter",
"bedGzLocation": {
"uri": "https://yourhost/sample_modkit.bed.gz"
},
"index": {
"location": {
"uri": "https://yourhost/sample_modkit.bed.gz.tbi"
}
}
}
}
JBrowse reads the score column (column 11 in bedMethyl, the methylation
fraction 0–1) and the name column (column 4, the modification code such as m
for 5mC or h for 5hmC) as the subtrack source label.
The COLO829 tumor modkit bedMethyl file is included in the demo config as the track "COLO829_tumor.ht_modkit.bed (as MultiQuantitativeTrack)" under the Methylation category (assembly hg38, chr21).