Multi-sample SV visualization w/ 1kG
This tutorial explores structural variants (SVs) from the 1000 Genomes Project using JBrowse's multi-sample visualization tools. We cover three connected analyses:
- Browsing population-level SV calls and their genotype distribution
- Examining SV inheritance in a parent–child trio
- Characterizing a large chromosomal inversion on chr19
For SNP-level trio analysis — phased genotypes, IBD blocks, and crossing-over visualization — see the companion Analyzing a phased trio tutorial.
Dataset
The 1000 Genomes Project sequenced genomes from 2,504 individuals across 26 populations. The 2022 high-coverage re-analysis produced a comprehensive SV callset (Byrska-Bishop et al., 2022) that includes deletions, insertions, inversions, and translocations with per-sample genotypes across all 2,504 individuals.
For this tutorial we use a pre-configured JBrowse instance that already has the SV callset and trio BAM tracks loaded. No data download is required.
Getting started
Open the 1000 Genomes demo instance and enable the variant track from the track selector (top-left menu icon of any linear view):
Under Variant calls in the track selector, enable the 1KGP 2022 Illumina ensemble SV callset. A track of orange SV bars will appear across the genome.
Browsing SVs with the SV inspector
The SV inspector combines a searchable/filterable table of all calls with a whole-genome circular overview. Open it from the menu bar: Add → SV Inspector view, then provide the VCF from the demo config (the URL is listed in the track's About track menu). The circular view renders inter-chromosomal translocations as orange chords; the table can be sorted and filtered by any column.
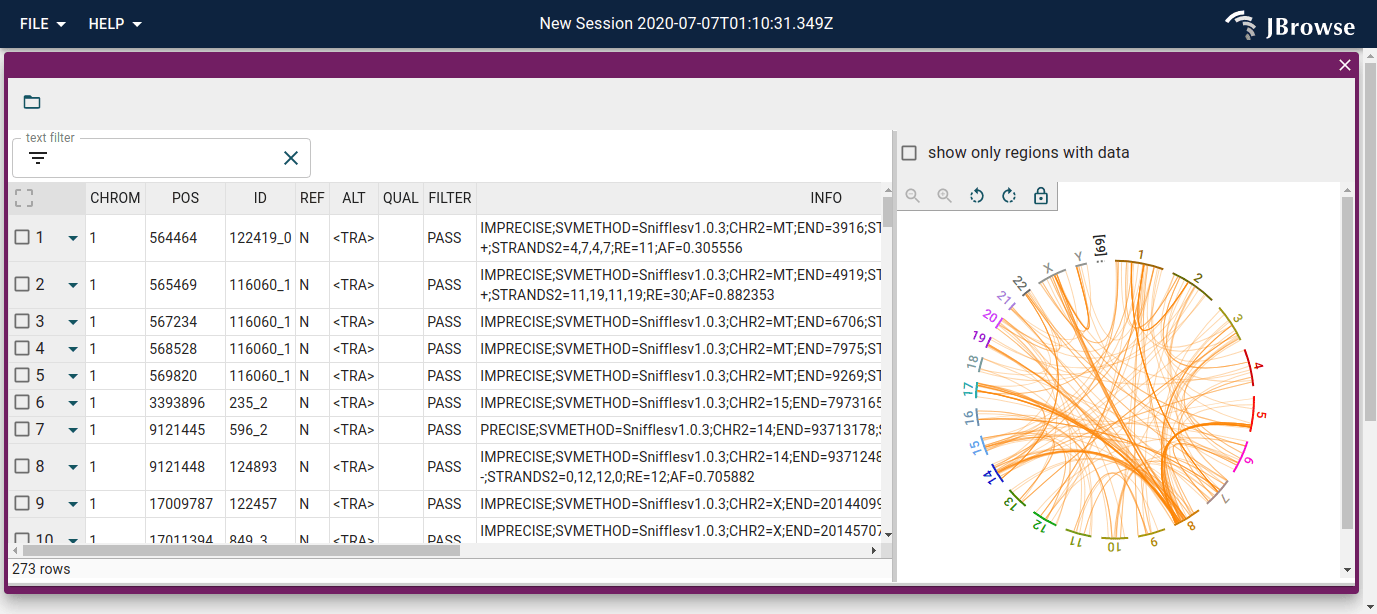
Useful filters to try:
- Type
DELin the SVTYPE column to isolate deletions - Type
INVto isolate inversions — these appear as arcs within a single chromosome arm rather than inter-chromosomal chords - Click any row to jump directly to that locus in the linear view
For a full walkthrough of loading data into the SV inspector, see the SV inspector guide.
Per-sample genotypes
With the SV track loaded in the linear view, click any variant bar to open its feature details panel. Scroll to the SAMPLES section — this lists every sample in the cohort with its genotype (GT), read depth, and other per-sample fields.
To see the genotype pattern across many SVs at once, switch display modes:
Track menu → Display types → Multi-sample variant display (regular)
This draws one row per sample in the track so you can scan across many variants simultaneously and spot which calls are private to a single sample, shared within a family, or present at high allele frequency across the cohort. See Multi-sample variant displays for details on the display modes.
Inspecting a trio
The demo includes BAM tracks for a parent–child trio, available under 1000 Genomes → Alignments in the track selector. Load all three (mother, father, child) to stack them beneath the variant track.
Open the extended trio session
Once you have an SV of interest, check the three trio rows in the SAMPLES table and the corresponding read tracks:
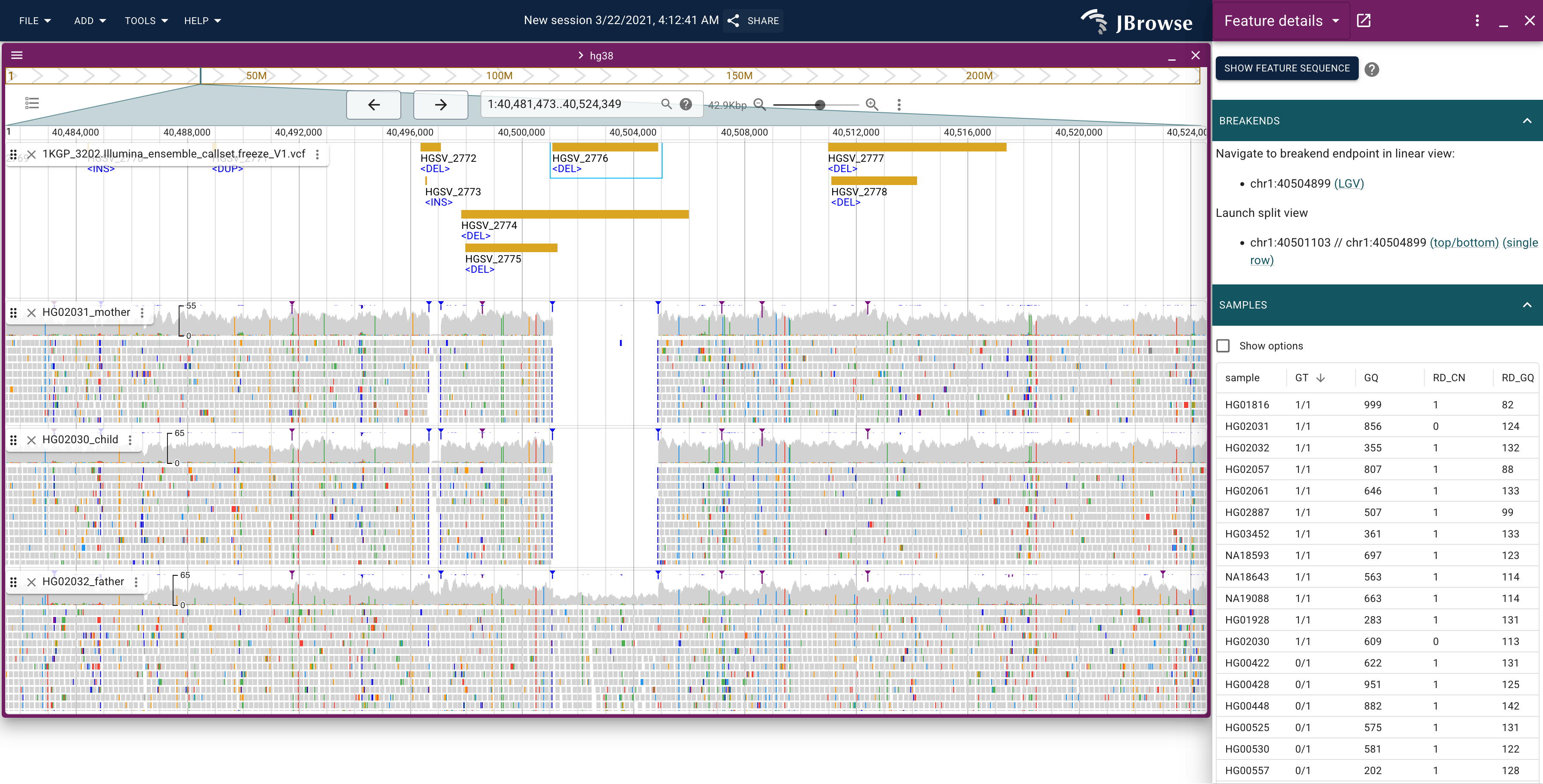
| Genotype pattern | Interpretation | | --------------------------- | -------------------------------------------- | | Child 0/1, both parents 0/0 | Candidate de novo SV | | Child 0/1, one parent 0/1 | Inherited from that parent | | Child 1/1, both parents 0/1 | Homozygous — inherited copy from each parent | | Child 0/0 | Not present in this individual |
The trio alignment tracks let you verify read-level support for each genotype. Look for coverage changes, soft-clipped reads, or orientation anomalies in each family member's track at the SV locus. For a primer on reading these signals, see the SV visualization guide.
For SNP-level trio phasing and IBD block analysis with the matrix display, see the Analyzing a phased trio tutorial.
The chr19 large inversion
The 1KGP SV callset includes a large well-studied inversion polymorphism on chromosome 19. Navigate to chr19:41,700,000–42,000,000.
At this scale the variant track shows the inversion call as a wide bar spanning the region. To separate samples by genotype, use:
Track menu → Cluster by genotype
This groups samples into ref/ref (0/0), het (0/1), and hom-alt (1/1) rows, making the population frequency of the inversion immediately visible — most samples are reference homozygous, with a subset carrying one or two copies of the inverted allele.
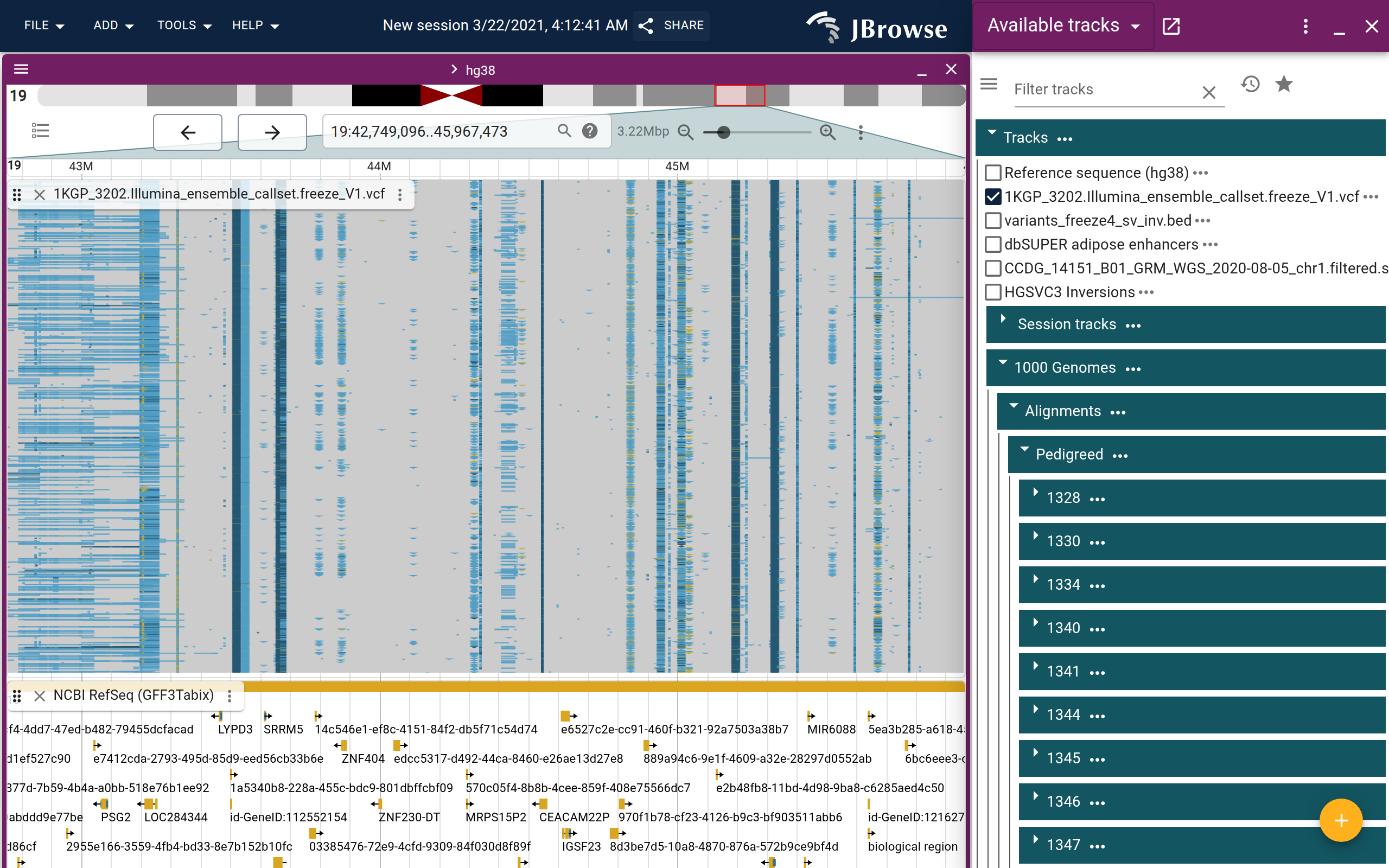
Open the inversion demo session
Read orientation evidence at the breakpoints
Zoom in to one of the inversion breakpoints (approximately chr19:41,749,000 or chr19:41,902,000) and enable pair orientation coloring on a BAM track:
Track menu → Pileup settings → Color by... → Pair orientation
At the breakpoint you will see:
- Teal (LL) pairs — both mates mapping to the forward strand — and dark blue (RR) pairs — both mates mapping to the reverse strand — clustering at the junction. These are the hallmark orientation signal of an inversion: reads that straddle the breakpoint change from the normal LR orientation to same- direction LL or RR.
- Soft-clipped reads at the exact breakpoint edge, where reads cannot align through the junction sequence
Switch to the Read arc display (Track menu → Display types → Read arc display) to see the long-range connections spanning the inversion. Arcs with LL/RR coloring that span the inverted interval confirm the rearrangement.
See the SV visualization guide — Inversion section for diagrams of these orientation patterns.
Breakpoint split view
Click the inversion bar in the variant track to open feature details. In the BREAKENDS section, click the split view link. This opens both inversion breakpoints side-by-side in synchronized panels, with splines connecting supporting reads across both panels and the variant call drawn as a colored line with directional feet.
For more on navigating the breakpoint split view, see Breakpoint split view.
Summary
| Step | Tool | What to look for | | ----------------------- | ------------------------------------------- | ------------------------------------------------------------ | | Population triage | SV inspector table + circular view | SV type counts; inter-chr translocations as chords | | Per-sample genotypes | Feature details → SAMPLES | GT 0/0 / 0/1 / 1/1 across all 2,504 samples | | Genotype patterns | Multi-sample display (regular) | High-frequency vs private calls; row pattern per sample | | Trio inheritance | Trio BAM tracks + SAMPLES table | De novo vs inherited; which parent contributed the alt | | Inversion genotyping | Cluster by genotype | Alt-genotype samples grouped into distinct rows | | Inversion read evidence | Pair orientation coloring; Read arc display | LL/RR pairs at breakpoints; long arcs spanning the inversion | | Breakpoint detail | Breakpoint split view | Splines + variant call across both junctions |
See also
- SV visualization guide — reference for all SV display types and SV-type read signatures
- SV inspector guide — loading data into the SV inspector
- Multi-sample variant displays — regular and matrix display mode details
- Analyzing a phased trio — SNP-level trio phasing and IBD block analysis
- Cancer Genome in a Bottle (SVs) — end-to-end SV workflow with a cancer dataset