Hi-C track
Hi-C captures genome-wide chromatin interaction frequencies by sequencing pairs of genomic regions that are physically co-located in the nucleus. JBrowse renders these as a triangular contact matrix where color intensity indicates contact frequency — brighter means more interactions.
JBrowse reads .hic files produced by Juicer, Juicebox, and compatible
pipelines, using the hic-straw module.
Loading a Hi-C track
In the Add track dialog, paste the URL to a .hic file (or open it from disk).
JBrowse auto-detects the format and creates a Hi-C track. The track renders a
triangular contact matrix below the chromosome ruler.
Reading the contact matrix
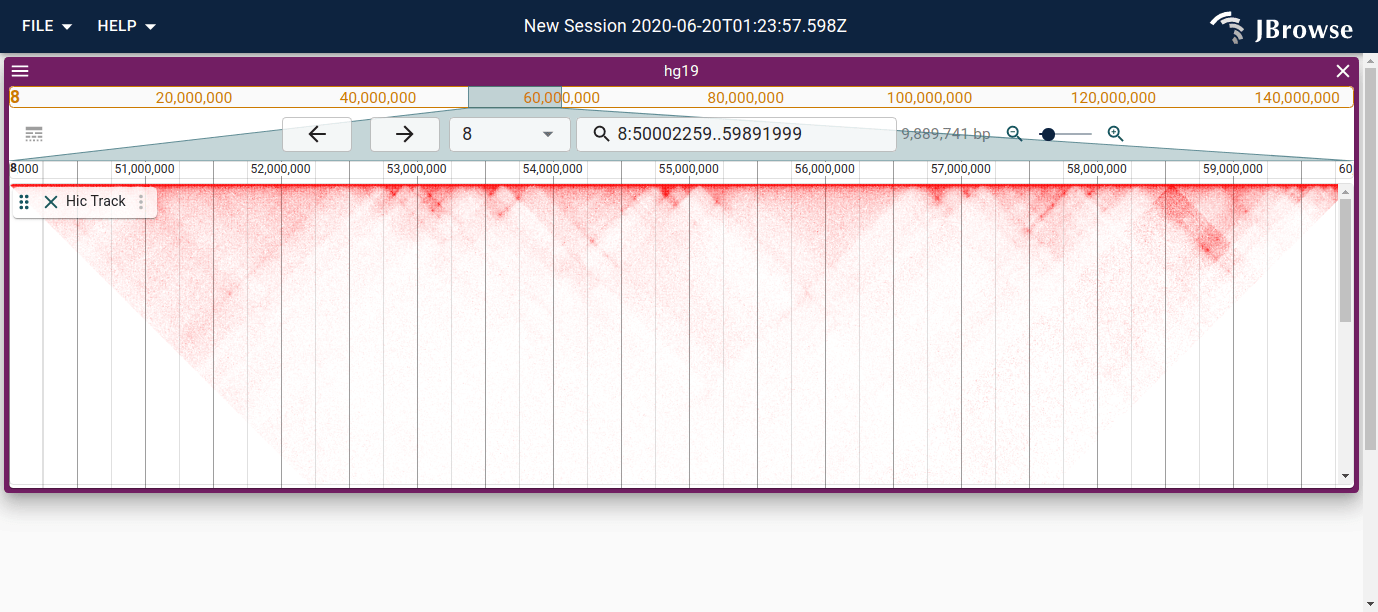
Key features to interpret:
- Diagonal — always the brightest band; self-interactions within a region
- TAD boundaries — sharp drops in signal that delimit the triangular blocks; the corners of these blocks are where loop anchors often occur
- Loops — punctate off-diagonal bright spots at specific locus pairs, typically CTCF-anchored
- Compartments — at low resolution, alternating plaid-like patterns (A/B compartments) reflecting active vs repressed chromatin
Adjusting resolution and color scale
JBrowse automatically selects a resolution that fits the current view width. Zoom in to increase resolution; zoom out to see larger-scale organization.
The color scale is configurable via the track's configuration — see the Hi-C track config guide for available options.
Live demo — Hi-C contact matrix with gene annotations