I'm pleased to announce the release of JBrowse 1.16.1! This release offers a variety of small bugfixes for cross-origin resource fetching, BAM files, and JBrowse desktop.
It also contains a fix that helps very large track lists to have a faster initial load for the Hierarchical track selector specifically.
Updated NeatCanvasFeatures to allow non-coding transcripts to be colored
differently with style->unprocessedTranscriptColor. Thanks to @billzt
for the bug report (issue #1298, @cmdcolin)
Bug fixes
Fixed issue where data directories with spaces in them were giving errors
due to CORS on JBrowse Desktop (issue #1285, @cmdcolin)
Fixed issue where the name store could be queried before being initialized
(issue #1286, @cmdcolin)
Fixed an issue for large BAM headers failing to load post-@gmod/bam
integration (@cmdcolin)
I am very pleased to announce the release of JBrowse version 1.16.0!
A major new feature of this release is the introduction of paired read visualization options!
And rather than delivering just one type of paired read visualization, there are many options for this including:
Pileup - standard alignments view but with connections between pairs
Read cloud - plotting by insert size
Arc view - plotting paired reads as connected arcs
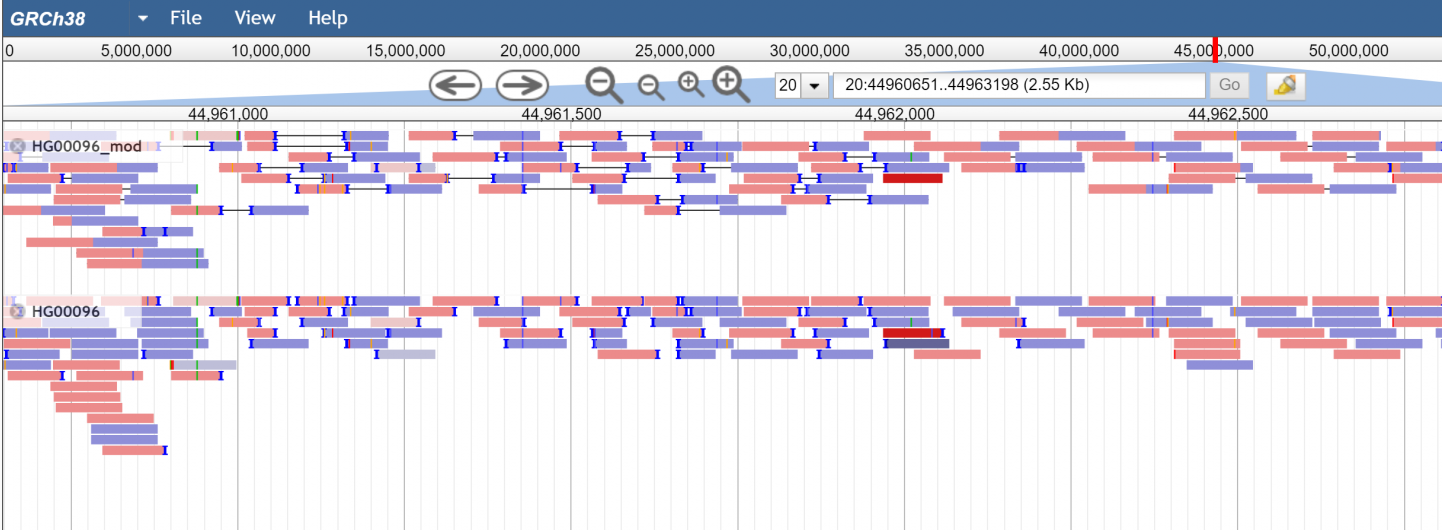
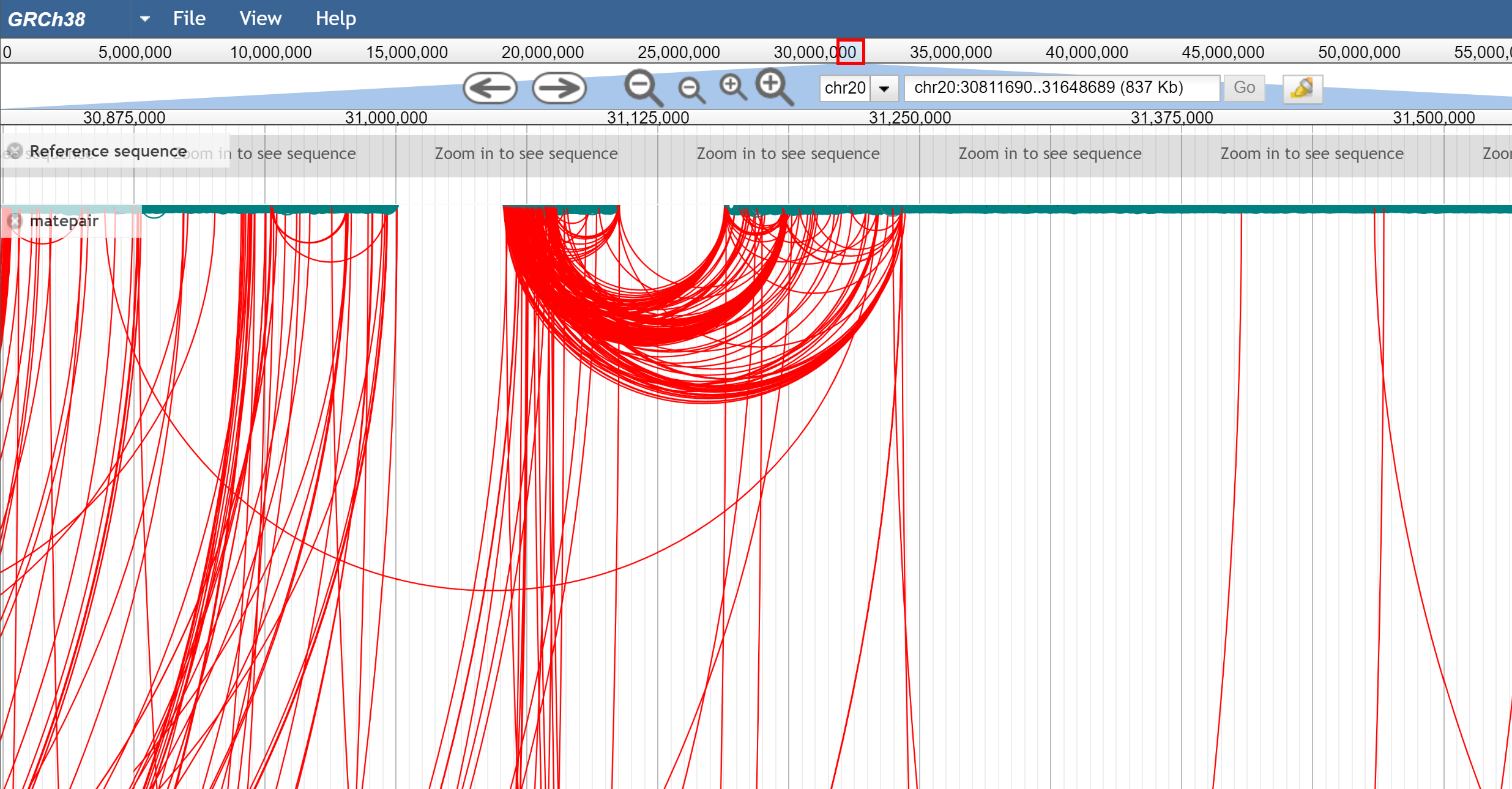
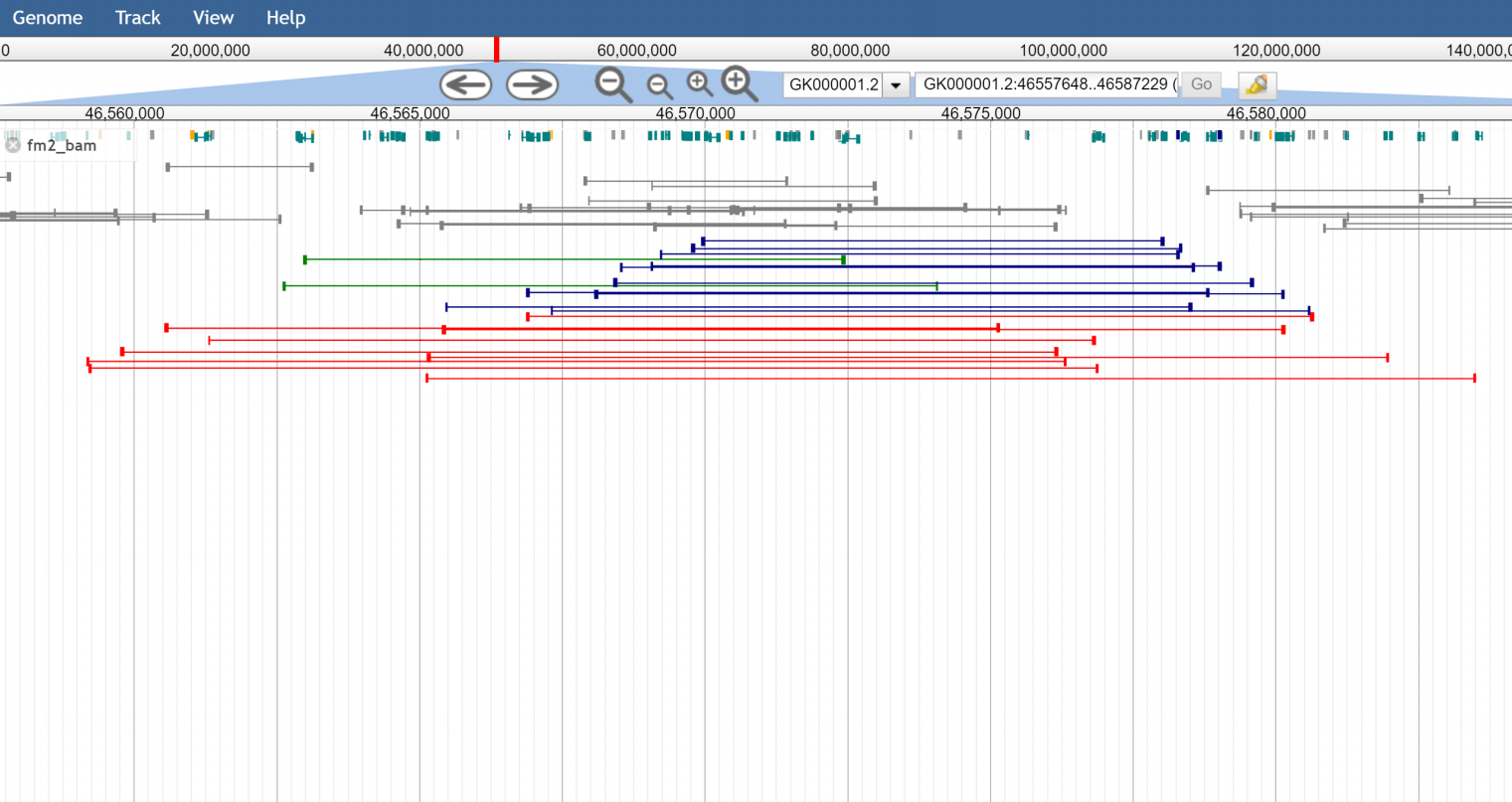
There are also multiple coloring options for each type of view, including coloring by insert size (so abnormally large inserts are colored red for example) or pair orientation. Here are some example screenshots to help demonstrate
Figure 1.standard pileup view with paired and unpaired modes on the same data.
Figure 2. arc view which shows many long-range connections from a mate pair dataset.
Figure 3. The read cloud view showing color by orientation and size (red being abnormally large insert size, and turquoise and blue being incorrect pair orientations)
JBrowse version 1.16.0 also contains many other features and bug fixes, so please review the changelog below! Also make sure to view the paired read documentation for more information on the paired read options.
Added ability to view paired read data as connected entities for BAM and
CRAM store classes. There are multiple different viewing options for this
including plotting by insert size, plotting as connected arcs, or pileup
views for the paired reads. Additonally multiple color schemes are available
for coloring by insert size, pair orientation, mapping quality, and more.
Thanks to @garrettjstevens, @rbuels, @AndyMenzies, and @keiranmraine for
testing. Also a big thanks to @jrobinso from @igvteam for contributions to
CRAM code related to paired reads (issue #1235, @cmdcolin)
Minor improvements
For users with the "dev" or compiling JBrowse from source code, the ./setup.sh
now performs a full webpack production build (issue #1223, @cmdcolin)
Created new BAM parsing mechanism using the npm module @gmod/bam.
Users might see some modest performance improvements due to enhanced tooling.
Thanks to @rbuels and others for testing and feedback (issue #1215, issue #1178,
@cmdcolin)
setup.sh now supports setting a JBROWSE_PUBLIC_PATH environment variable for
more flexibility in iframeless embedding scenarios (issue #1213, @rbuels)
Added support for indexing arbitrary fields from GFF3Tabix files by setting
nameAttributes in the track config e.g. nameAttributes=name,id,customfield.
(issue #1115, issue #1222, @cmdcolin)
Add support for generate-names to index VCF features that have multiple IDs in
the ID column (@cmdcolin)
Added documentation on embedding JBrowse in an iframe and in a div, including
how to embed JBrowse using a custom JavaScript object as a configuration
(issue #1228, issue #1243, @rbuels and @garrettjstevens)
Added ability to render non-coding transcript types to the default Gene glyph
which helps when a gene feature has a mix of coding and non-coding subfeatures
(issue #1106, issue #1230, @cmdcolin)
Added a config datasetLinkToParentIframe to make the dataset selector use
window.parent for when jbrowse is in an iframe (issue #1248, @enuggetry)
Improved error message that is displayed when a data file cannot be fetched
via CORS (@rbuels)
Added some word wrapping for long unbroken fields in the View details
popups. Thanks to @luke-c-sargent for the idea (issue #1246).
Added hideImproperPairs filter for Alignments2/SNPCoverage tracks which
disambiguates from missing mate pairs (issue #1235, @cmdcolin)
Added useTS coloring option for RNA-seq strandedness, similar to the useXS
that existed previously (issue #1235, @cmdcolin)
Added a --bgzip_fasta option for prepare-refseqs.pl and also the ability
to index reference sequence names if they are manually specified as a
fasta index e.g. refSeqs=genome.fai. Thanks to @FredericBGA for the report!
(issue #1281, issue #1282, @cmdcolin).
Fixed issue with getting feature density from BAM files via the index stats
estimation (issue #1233, @cmdcolin)
Fixed issue where some feature mouseovers where not working properly (issue
#1236, @cmdcolin)
Fixed issue where instantiating JBrowse via standalone.js didn't work when
in a production build with JBROWSE_PUBLIC_PATH overridden (issue #1239,
@garrettjstevens)
Small fix for issue where SNPCoverage would crash on some feature filters
(issue #1241, @cmdcolin)
Fixed issue where JBrowse Desktop was not able to access remote files (issue
#1234, issue #1245, @cmdcolin)
Fixed bug where some files were not being fetched properly when changing
refseqs. Thanks to @luke-c-sargent for the report (issue #1252)
Fixed storeTimeout on CRAM files being unused which can result in excessive
fetches (issue #1235, @cmdcolin)
Fixed issue where JBrowse would load the wrong area of the refseq on startup
resulting in bad layouts and excessive data fetches. Thanks to @hkmoon,
@cmdcolin, and @garrettjstevens for debugging (issue #1190, issue #1235, pull
#1187)
Fixed issue where CRAM layout and mouseover would be glitchy due to ID
collisions on features (issue #1271, @cmdcolin)
Fixed issue where some parts of a CRAM file would not be displayed in JBrowse
due to a CRAM index parsing issue (@cmdcolin)
Fixed an issue where BAM features were not lazily evaluating their tags
(@cmdcolin)
Notable changes
Rendering of features in popups, mouseover tooltips, and feature labels were
made to escape HTML. If you are using literal HTML labels in these places
then set the attribute unsafePopup, unsafeMouseover, or unsafeHTMLFeatures
on your tracks. Thanks to @garrettjstevens (issue #1263, @cmdcolin).
JBrowse 1.15.4 has been released! This release allows you to use bgzip indexed FASTA, adds a new categoryOrder config for sorting the Hierarchical track selector, and more!
Thanks to everyone for their feature requests and bug reports, and enjoy!
Added support for bgzipped indexed FASTA. To use, bgzip your FASTA with
bgzip -i file.fa, which generates file.fa.gz and file.fa.gzi and then use
samtools faidx file.fa.gz. If you specify the .fa.gz in the track config e.g.
"urlTemplate": "file.fa.gz" and have all three files in your data directory,
then they will automatically be detected (issue #1152, issue #1200, @cmdcolin)
Allow fna and mfa file extensions for FASTA to be recognized by default in
the Open sequence dialog (issue #1205, @cmdcolin)
Added a topLevelFeaturesPercent configuration variable that can be used to
correct feature statistics estimates when topLevelFeatures is being used for
a track, or when it contains deeply-nested features. This configuration variable
is currently only used by BAM, BEDTabix, GFF3Tabix, and VCFTabix stores.
(issue #1147, issue #1209, @rbuels)
Tabix-based data stores use a new storage backend based on the @gmod/tabix npm
module. Users should see some modest performance improvements for Tabix-based
tracks. (issue #1195, issue #1209, @rbuels)
Added categoryOrder config to allow sorting the categories in the Hierarchical
track selector. For example, categoryOrder=VCF,Quantitative/Density,BAM. Note
that we specify a lowest level subcategory e.g. Quantitative/Density to sort the
parent category Quantitative to a position (issue #1203, issue #1208, @cmdcolin)
Bug fixes
Fixed a bug in which feature labels would sometimes be repeated across the view,
in the wrong locations. (@rbuels)
Fixed error where a chunk size limit error during histogram display would not be
displayed (@cmdcolin)
Fixed issue where Open sequence dialog will open up the default "data" directory
instead of a blank instance (issue #1207, @cmdcolin)
Added check for PCR duplicates for CRAM features (@cmdcolin)
Fixed issue where editing the track names and types in the "Open track" dialog box
was not working when editing multiple tracks (issue #1217, @cmdcolin)
Fixed issue in which large VCF headers were not always correctly parsed by JBrowse
(issue #1139, issue #1209, @rbuels)
Fixed issue where the histogram Y-scale bar would appear over features (issue
#1214, issue #1218, @cmdcolin)
JBrowse 1.15.3 has been released! This contains an bugfix introduced in 1.15.0 where typing a reference sequence name in the search bar did not navigate to it properly.
It also contains a cool feature where "ultra minimal configs" can be specified by simply specifying a track label and urlTemplate
For example, a directory containing indexed files like VCF, BAM, GFF3 tabix, and indexed FASTA can have a config simply like this in tracks.conf
Add ability to automatically deduce the storeClass and trackType of files based on
the file extension of urlTemplate. This allows very minimal configs where only
track label and urlTemplate can be specified. (issue #1189, @cmdcolin)
Bug fixes
Fixed an issue with servers that use HTTP Basic Authentication on certain browsers,
notably some Chromium, Firefox 60 and earlier, and Safari. Thanks to Keiran Raine
for reporting and @cmdcolin for debugging. (issue #1186, @rbuels)
Fix issue where searching for reference sequence names would not be navigate to the
typed in reference sequence (issue #1193, @cmdcolin)
JBrowse 1.15.2 has been released! This contains a couple nice features including
estimating feature density directly from the index file of BAM, VCF, and other
tabix formats. We also updated the website with a new blogging and documentation
platform! Please try it out and give us feedback.
Created "index stats estimation" which overrides the older "global stats estimation"
that randomly samples genomic regions of BAM, VCF, etc to find feature density. This
allows initial track load to be faster automatically. (issue #1092, issue #1167,
@cmdcolin)
Removed the "full" or "dev" releases from the build. If you need a "dev" release, you
can simply download the JBrowse "source code" link from the GitHub releases page, or
use a git clone of the JBrowse repository. This will behave the same as the "dev"
release. (issue #1160, issue #1170, @cmdcolin)
Updated the main jbrowse.org website to use the docusaurus platform. The main docs
for the website are now moved from the GMOD.org server to jbrowse.org. You can find
the latest documentation in the header bar. We hope you will enjoy this upgrade!
There is also a new quick start guide based on setting up JBrowse with indexed file
formats. (issue #1153, issue #1137, issue #1173, @cmdcolin)
Bug fixes
Added a more robust HTML processing in Util.js. Thanks to @hkmoon for the idea and
implementation. (issue #1169, @hkmoon)
Fixes issue where navigating away from genome browser and returning would not remember
the location. Thanks to Vaneet Lotay for reporting. (issue #1168, @cmdcolin)
Fixes off-by-one in the display of the size of the genomic region being viewed. Thanks
to @sammyjava for the bug report! (issue #1176, @cmdcolin)
Add a internal code attribute for XHR requests that use byte-range headers so that if a
server does not support it, an error is returned immediately. Thanks to @theChinster
for the motivating example (issue #1131, issue #1132, issue #1134, @cmdcolin).
Speed up TwoBit file processing with a robust implementation of the file spec. The
improvements are contained in a new npm module @gmod/twobit.
Thanks to @cmdcolin for some testing and motivating examples. (issue #1116, issue #1146,
@rbuels)
Added feature.get('seq') to CRAM features which enables detailed comparison of the
read versus the reference with the renderAlignment configuration. (issue #1126,
issue #1149, @rbuels)
Added support for 1000genomes CRAM 2.0 codecs via updates to the @gmod/cram npm module.
(@rbuels)
Add some better formatting for rich metadata in the "About this track" dialog boxes for
tracks. Thanks to Wojtek Bażant for the idea and implementation! (issue #1148, @wbazant)
Bug fixes
Fix bug where prepare-refseqs with indexed FASTA would allows scrolling past the end of
the chromosome (@cmdcolin).
Fix long standing bug related to not being able to configure dataRoot in the config file.
Now you can set dataRoot=mydirectory to make JBrowse load mydirectory instead of the
default data by default (issue #627, issue #1144, @cmdcolin).
Added hashing of the BAM feature data to generate unique IDs in order to distinguish
reads that have nearly identical information (same read name, start, end, seq, etc).
If the reads literally have identical information in them JBrowse is still unable to
display but this generally seems to be due to limited use case such as secondary
alignments in RNA-seq (issue #1108, issue #1145, @cmdcolin)
JBrowse 1.15.0 has been released! This is a really big one.
At long last, JBrowse can open and view CRAM v2 and v3 files just like BAM. In fact, viewing CRAM files can sometimes be even faster than viewing BAM files, because there is less data to move around! Enjoy CRAM support, we all worked really hard to bring it to you! Getting started with CRAM is easy, just use the JBrowse/Store/SeqFeature/CRAM store and the Alignments2 track type, just as you probably are already with BAM files. It works with local files too, of course!
Another big development, JBrowse Desktop has matured to the point where we can recommend it without reservation to those looking for a fast, easy to use desktop genome browser for local and remote files. From now on, we'll be publishing builds of JBrowse Desktop for Windows, Mac OS, and Linux alongside the regular JBrowse releases. Download it and give it a try, let us know what you think! Huge congratulations to Colin Diesh (who is now a full-time JBrowse developer!) for thinking of this, and seeing this amazing development through. I think you will be quite pleased with how well JBrowse runs on the desktop!
Also, JBrowse now supports CSI format indexes for BAM fiels and Tabix-indexed (VCF, GFF3, etc) files. Now your BAM files can be even bigger. As if they weren't huge enough already. This continues our very serious commitment to make JBrowse effortlessly usable on even the biggest datasets.
Lastly, JBrowse now sports a nice new text-searching interface accessible from the View → Search menu item in the top bar. This was a cool idea that came out of discussions last month at the GCCBOSC 2018 CollaborationFest, and was executed with alacrity by Colin!
As always, read on below the fold for the full release notes, including minor improvements and bugfixes. And thanks for using JBrowse. 😁
Added support for the CSI index format for tabix VCF/BED/GFF and BAM files! This allows
individual chromosomes longer than ~537MB (229 bases) to be used in JBrowse. To enable,
use the csiUrlTemplate config to point to the file. The "Open track" dialog also allows
CSI to be used. Thanks to Keiran Raine for initial report and Nathan S Watson-Haigh for
catching a bug in the initial implementation! (issue #926, issue #1086, @cmdcolin)
Added a new search dialog box via the View->Search features menubar. It will search the
currently configured store for features. You can also configure the dialog class in the
configuration with names.dialog entry, or disable search dialog with disableSearch.
Thanks to the #GCCBOSC hackathon for the idea and feedback (issue #1101, @cmdcolin).
Minor improvements
Re-enabled JBrowse Desktop builds for releases! The Windows, Mac, and Linux binaries for
JBrowse Desktop are uploaded automatically to GitHub releases page. JBrowse Desktop is a
standalone app that can be used without a web server, similar to IGV or IGB (@cmdcolin)
Added a dontRedispatch option for GFF3Tabix stores. Example: set dontRedispatch=region
if there are very large region biotype features in the GFF that do not have subfeatures which will
speed up loading times significantly (issue #1076, issue #1084, @cmdcolin)
Add auto-lower-casing to the feature.get('...') function, commonly used for callback
customizations. Now, for example, feature.get('ID') works as well as feature.get('id').
Thanks to @nvteja for motivating this! (issue #1068, issue #1074, @cmdcolin)
Added cache-busting for track config files which actively prevents stale configuration files
from being loaded (issue #1080, @cmdcolin)
Added indexing of both Name and ID from GFF3Tabix files from generate-names.pl. Thanks to
@billzt for the implementation! (issue #1069)
Made the color of the guanine (G) residue more orangey than yellow to help visibility.
Thanks to Keiran Raine for the implementation! (issue #1079)
Refactored NeatCanvasFeatures and NeatHTMLFeatures as track types. You can enable the track
style on specific tracks instead of globally this way by modifying the track type to be
NeatCanvasFeatures/View/Track/NeatFeatures or NeatHTMLFeatures/View/Track/NeatFeatures.
(issue #889, @cmdcolin).
In the location box, allow strings with format ctgA:1-100 e.g. with a hyphen instead of ...
Big thanks to Nathan S Watson-Haigh for the idea and implementation! The default display
remains .. but - is allowed. (issue #1100, issue #1102, @nathanhaigh)
Allow sequences with a colon in their name to be used in the location box. This includes
the HLA reference sequences in hg38. Thanks again to Nathan S Watson-Haigh for the
implementation of this feature. (issue #1119, @nathanhaigh)
Fix sensitivity to .gff.gz vs .gff3.gz in GFF3Tabix tracks opened via the "Open track"
dialog for GFF3Tabix. (issue #1125, @cmdcolin)
Feature detail dialog boxes now display subfeatures of features on the reverse strand in
upstream-to-downstream order, instead of in genomic coordinate order. Thanks to
@nathanhaigh for suggesting this and contributing the fix! (issue #1071, issue #1114, @nathanhaigh)
Bug fixes
Fixed a potential cross-site-scripting (XSS) vulnerability by disallowing dataRoot config
values or ?data= URL parameters that point to a different server from the one serving
JBrowse. Users can disable this security check by setting allowCrossOriginDataRoot = true
in their configuration. (@cmdcolin, @rbuels)
Fixed a memory leak that was introduced in JBrowse 1.13.1 in generate-names.pl. Thanks to
@scottcain for reporting (issue #1058, @cmdcolin)
Fix the error checking in setup.sh if no node is installed at all (issue #1083, @cmdcolin)
Fix calculation of histograms on GFF3 and GFF3Tabix stores. Thanks to @thomasvangurp for
the bug report and sample data! (issue #1103, @cmdcolin)
Fix the representation of array-valued attributes in column 9 for GFF3Tabix. Thanks to
@loraine-gueguen for the bug report! (issue #1122, @cmdcolin)
Fixed a bug in which visibleRegion() in GenomeView.js sometimes returned a non-integer value for end, which interfered with some scripts and plugins. Thanks to @rdhayes for noticing and contributing the fix! (issue #491, @rdhayes)
Fixed bug where reference sequences with names containing the : character could not be switched to by typing their name in the search box. (issue #1118, issue #1119, @nathanhaigh)
Added a datasetSelectorWidth configuration key that sets the width of the dataset
selector. The width defaults to 15em. Example setting in tracks.conf:
[GENERAL] classicMenu = true datasetSelectorWidth = 20em
Thanks to @srobb1 for pointing out the need for this. (issue #1059, @rbuels)
When exporting GFF3 from the 'Save track data' menu, the ##sequence-region pragma now
specifies the exact sequence region that was exported from the UI. Thanks to @mwdavis2
for pointing this out! (issue #905, @rbuels)
Fixed the --config option for add-bw-track.pl. Although documented in the script's POD,
it was not actually being processed. Thanks to @loraine-gueguen for noticing it, and for
contributing the fix! (issue #1063, issue #1064, @loraine-gueguen)
Fixed a bug in which setup.sh failed if run twice in a row under some circumstances.
(issue #1053, @cmdcolin)
Fixed a bug in which setup.sh did not accept nodejs version 10 as sufficiently recent.
(issue #1048, @cmdcolin)
Fixed a bug in which the "Loading..." message erroneously appeared at the top of the
dataset-selection page. Many thanks to @srobb1 for noticing this and reporting it!
(issue #1057, @rbuels)
JSON syntax errors in the new configuration loading code now have better error messages.
Thanks to @billzt for pointing out the need for this! (issue #1061, @rbuels)
JBrowse now supports .idx indexes for VCFs that are generated by igvtools or GATK. Currently
only VCF files can be used with this index type, but this could be expanded to other file types
if users are interested. Thanks to @thon-deboer for suggesting this! (issue #1019, @rbuels)
The dropdown dataset selector in "classic menu" mode is now a type-ahead combo box, enabling
fast searching through large numbers of datasets. Thanks to @keiranmraine for the suggestion!
(issue #752, @rbuels)
There is now a new event named /jbrowse/v1/n/tracks/redrawFinished that fires after the
view is refreshed, when all of the visible tracks are finished drawing (or have errored).
Thanks to @scottcain for suggesting this. (issue #1027, @rbuels)
Improve the calculation of feature density for GFF3Tabix and add new one for GFF3 in-memory.
Thanks to @hkmoon for the suggestion! (issue #1039, issue #913, @cmdcolin)
Re-enabled JBrowse Desktop builds based on automatically building on Travis-CI
(issue #1028, @cmdcolin)
Bug fixes
Fixed several bugs related to the file-opening dialog's handling of indexed file types
(bam+bai, gz+tbi, etc). Thanks to @sletort for submitting the bug report! (issue #1033, @rbuels)
The Perl formatting tools now properly read include-ed configuration files. Thanks to @carrere
for pointing out this bug. (issue #551, @rbuels)
Fixed a bug in which the faceted track selector was nonfunctional in Internet Explorer 11.
(issue #1036, @rbuels)
This release includes two major improvements, the first being that it's no longer strictly necessary to run JBrowse inside an <iframe> when embedding it in another site or webpage. Big thanks to Lacey Sanderson for championing this feature, and we look forward to working with the Tripal team toward a better JBrowse integration!
The second major improvement is in displaying data from BigBed files. Not only can JBrowse can now open BigBed files both locally, and over the web, but it now natively displays BED-type thick/thin features and feature blocks rather than converting them to GFF3-style feature hierarchies. This makes both BigBed and BED fast and responsive. The new BED glyph will also use each feature's RGB color if it has one.
There are many more features coming that enhance JBrowse's compatibility with UCSC-format data, stay tuned for future JBrowse releases!
As always, read on below the fold for the full release notes, including minor improvements and bugfixes.
JBrowse now behaves much better when embedded in a webpage without using an iframe.
See tests/drupal.htm in the JBrowse code for an example of this usage. Thanks to
@laceysanderson for her patiently championing this feature all the way through the
long road to completion! (issue #777, issue #844, @cmdcolin)
There is a new BigBed store type, for opening BigBed files. An example BigBed track configuration:
JBrowse now has much better support for UCSC-style BED and BigBed features, via the new BED
glyph type for CanvasFeatures. Rather than rendering a complex feature hierarchy like many
of the other CanvasFeatures glyphs, the BED glyph draws sub-blocks with thick and thin regions,
for compatibility with the UCSC browser. CanvasFeatures will automatically use the
JBrowse/View/FeatureGlyph/UCSC/BED glyph type if a feature has no subfeatures, but has
blockCount or thickStart attributes. This means that, in practice, a BigBed file will
display very well with just the default configuration. Also for compatibility with the UCSC
browser, JBrowse will set a BED feature's background color if one is included in the
feature data (turn this off by setting itemRgb = false).
Minor improvements
The current dataset name is now displayed in the top right portion of the menu bar.
(issue #767, @rbuels)
prepare-refseqs.pl now accepts a --gff-sizes option to allow defining reference
sequence sizes from the ##sequence-region directives in a GFF3 file. @rbuels
Some store types now support a topLevelFeatures configuration variable, which allows
tracks to treat certain types of features as 'top-level', even the actual track data
has them as children of other features. One common use case for this would be if
you have gene models in a GFF3 structured as gene→mRNA→exon/CDS/UTR, but you want to
display the "mRNA" features as top-level, i.e. ignore the gene container that they are in.
Now you can set topLevelFeatures = mRNA in the track configuration, and the track will
display only "mRNA" features on the top level, ignoring any other existing top-level features,
and ignoring the containing "gene" features. This helps address what seems to be a common
pain point of having to "filter" tabix-formatted GFF3 before using it with Apollo. One
important caveat is that users that configure tracks to use an "out of band" source of
feature density or coverage data, like a separate wiggle file that shows feature density,
will have to make sure that the density data is correct for this filter setting if they
use it. Thanks to @Yating-L, @nathandunn, and @cmdcolin for valuable discussions.
Stores that support topLevelFeatures currently are: GFF3Tabix, GFF3, BED, BEDTabix,
GTF, and REST (issue #974, issue #969, @rbuels)
JBrowse can now accept additional configuration from a data-config attribute on its
container element. This is useful for embedding JBrowse in other sites, particularly
in cases where the JBrowse assets and configuration are stored or referenced from a
different location from the page displaying the embedded JBrowse. For example:
would tell JBrowse to look for its configuration and assets at the relative base URL
"../jbrowse". [@rbuels](https://github.com/rbuels)
* JBrowse now has a favicon! ([issue #973](https://github.com/gmod/jbrowse/issues/973), [@rbuels](https://github.com/rbuels))
* Added additional caching code to SequenceChunks and NCList stores, reducing duplicate
network requests and increasing performance in some circumstances ([issue #855](https://github.com/gmod/jbrowse/pull/855), [@cmdcolin](https://github.com/cmdcolin))
Bug fixes
Fix a bug in which saving exported data to a file was nonfunctional for some export
data types. @rbuels
Fix a bug in which subfeatures were not always fetched correctly when using the GFF3Tabix
store (issue #780, @rbuels)
Fixed several bugs with specific cases of relative URLs used in configuration. @rbuels