JBrowse 1.16.0 maintenance release
I am very pleased to announce the release of JBrowse version 1.16.0!
A major new feature of this release is the introduction of paired read visualization options!
And rather than delivering just one type of paired read visualization, there are many options for this including:
- Pileup - standard alignments view but with connections between pairs
- Read cloud - plotting by insert size
- Arc view - plotting paired reads as connected arcs
There are also multiple coloring options for each type of view, including coloring by insert size (so abnormally large inserts are colored red for example) or pair orientation. Here are some example screenshots to help demonstrate
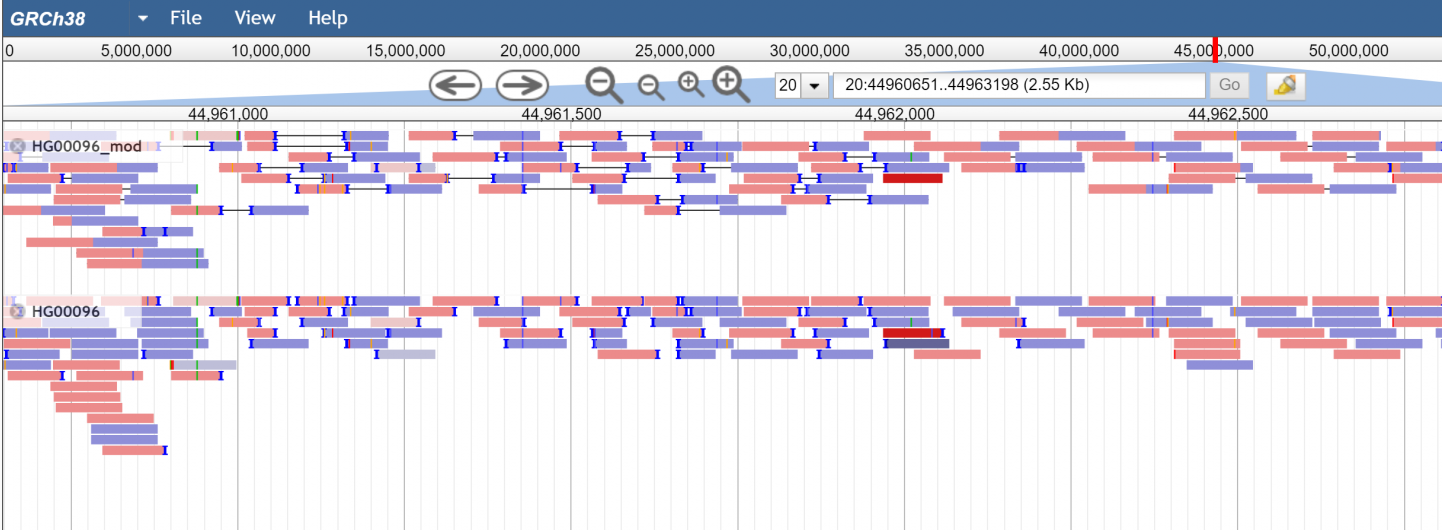
Figure 1.standard pileup view with paired and unpaired modes on the same data.
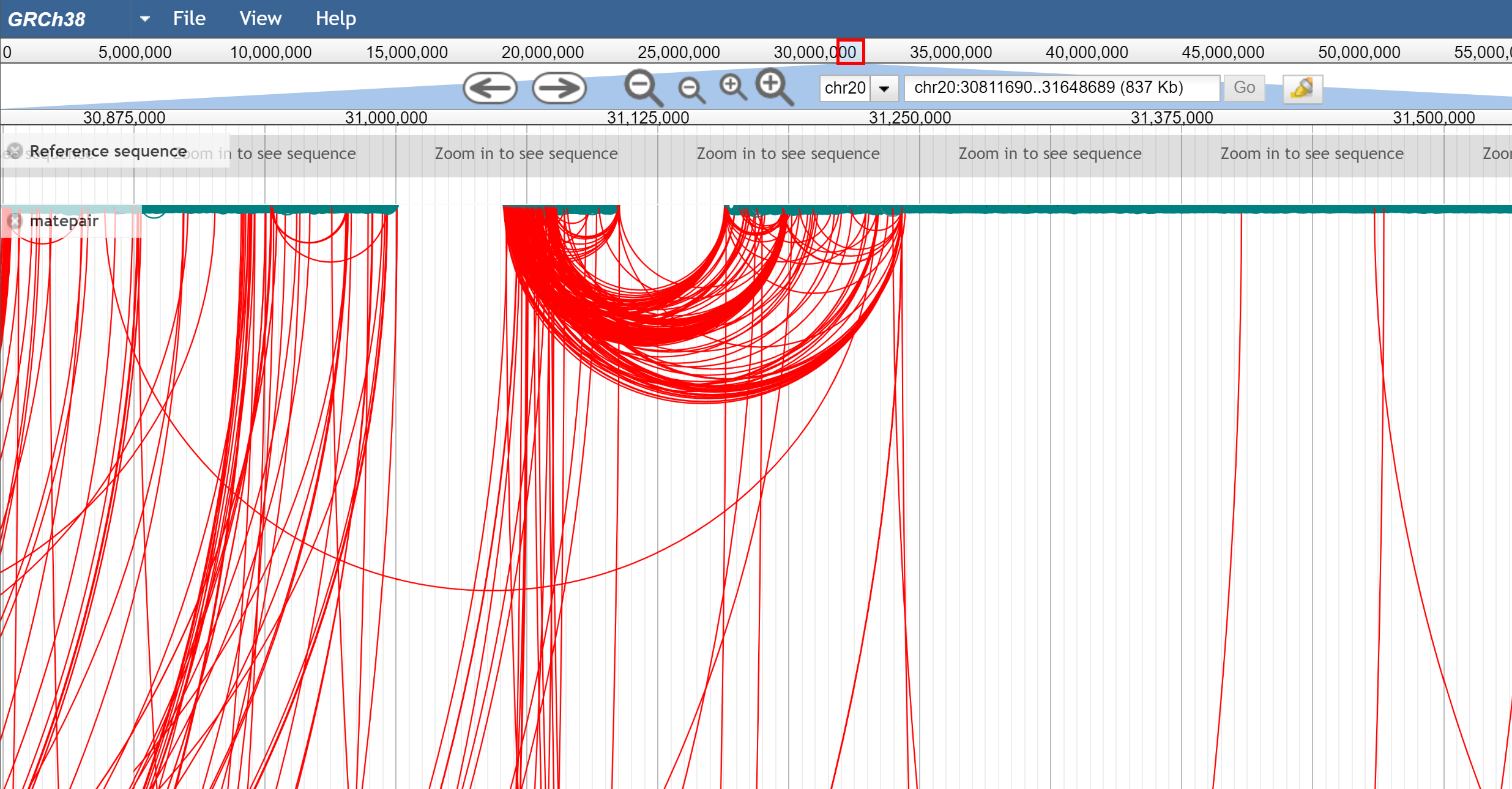
Figure 2. arc view which shows many long-range connections from a mate pair dataset.
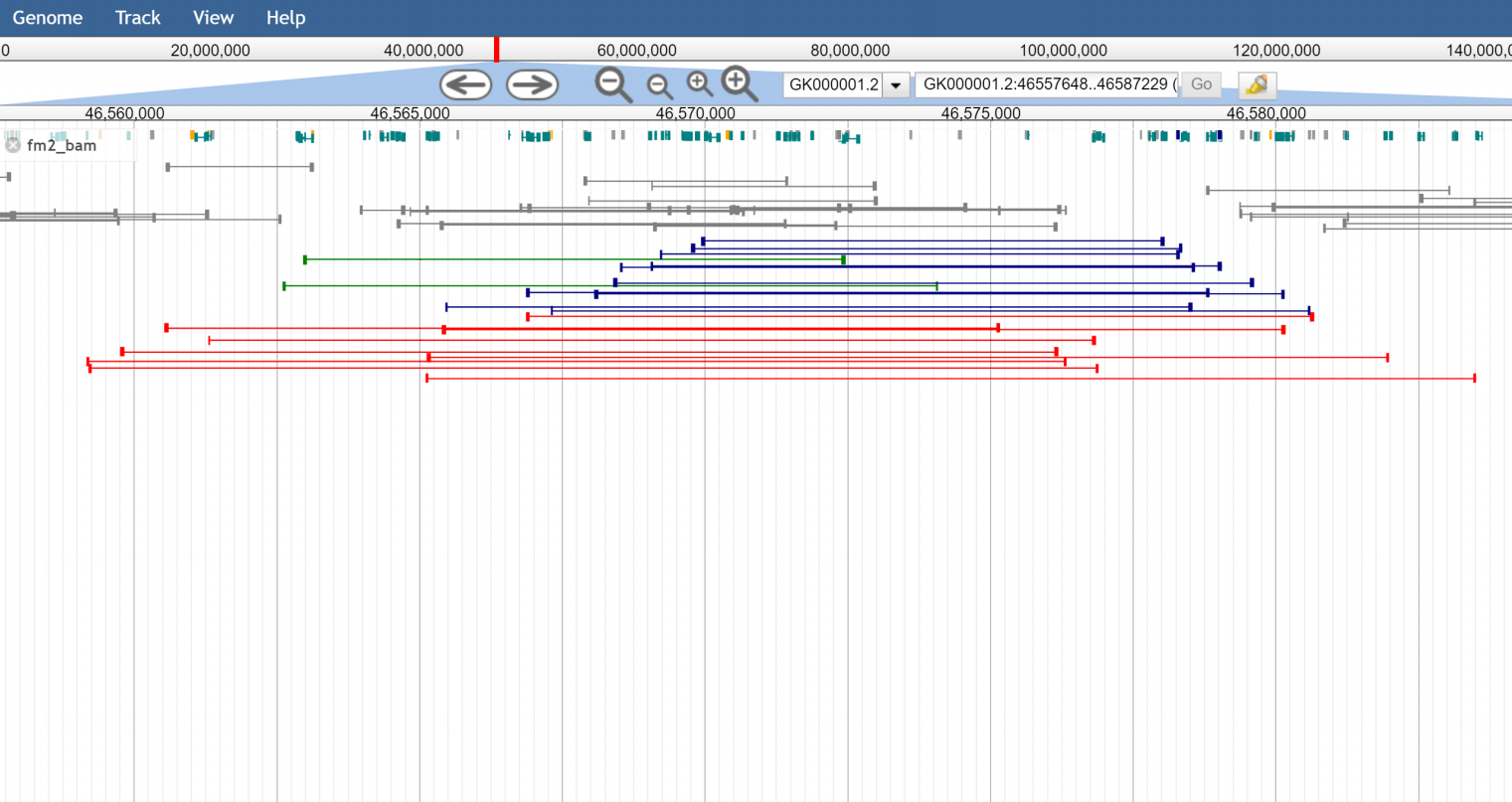
Figure 3. The read cloud view showing color by orientation and size (red being abnormally large insert size, and turquoise and blue being incorrect pair orientations)
JBrowse version 1.16.0 also contains many other features and bug fixes, so please review the changelog below! Also make sure to view the paired read documentation for more information on the paired read options.
- JBrowse-1.16.0.zip - minified release
- JBrowse-1.16.0-dev.zip - use this if you modify jbrowse source code or use plugins
- JBrowse-1.16.0-desktop-win32-x64.zip
- JBrowse-1.16.0-desktop-linux-x64.zip
- JBrowse-1.16.0-desktop-darwin-x64.zip
Major improvements
- Added ability to view paired read data as connected entities for BAM and CRAM store classes. There are multiple different viewing options for this including plotting by insert size, plotting as connected arcs, or pileup views for the paired reads. Additonally multiple color schemes are available for coloring by insert size, pair orientation, mapping quality, and more. Thanks to @garrettjstevens, @rbuels, @AndyMenzies, and @keiranmraine for testing. Also a big thanks to @jrobinso from @igvteam for contributions to CRAM code related to paired reads (issue #1235, @cmdcolin)
Minor improvements
For users with the "dev" or compiling JBrowse from source code, the ./setup.sh now performs a full webpack production build (issue #1223, @cmdcolin)
Created new BAM parsing mechanism using the npm module @gmod/bam. Users might see some modest performance improvements due to enhanced tooling. Thanks to @rbuels and others for testing and feedback (issue #1215, issue #1178, @cmdcolin)
setup.sh now supports setting a
JBROWSE_PUBLIC_PATHenvironment variable for more flexibility in iframeless embedding scenarios (issue #1213, @rbuels)Added support for indexing arbitrary fields from GFF3Tabix files by setting nameAttributes in the track config e.g. nameAttributes=name,id,customfield. (issue #1115, issue #1222, @cmdcolin)
Add support for generate-names to index VCF features that have multiple IDs in the ID column (@cmdcolin)
Added documentation on embedding JBrowse in an iframe and in a div, including how to embed JBrowse using a custom JavaScript object as a configuration (issue #1228, issue #1243, @rbuels and @garrettjstevens)
Added ability to render non-coding transcript types to the default Gene glyph which helps when a gene feature has a mix of coding and non-coding subfeatures (issue #1106, issue #1230, @cmdcolin)
Created new VCF parsing mechanism using the NPM module @gmod/vcf. Thanks to @cmdcolin and others for testing and feedback (issue #1227, issue #1199, @garrettjstevens)
Added ability to open "chrom.sizes" files from the Open sequence dialog (issue #1250, issue #1257, @cmdcolin)
Added a config
datasetLinkToParentIframeto make the dataset selector use window.parent for when jbrowse is in an iframe (issue #1248, @enuggetry)Improved error message that is displayed when a data file cannot be fetched via CORS (@rbuels)
Added some word wrapping for long unbroken fields in the View details popups. Thanks to @luke-c-sargent for the idea (issue #1246).
Added
hideImproperPairsfilter for Alignments2/SNPCoverage tracks which disambiguates from missing mate pairs (issue #1235, @cmdcolin)Added
useTScoloring option for RNA-seq strandedness, similar to theuseXSthat existed previously (issue #1235, @cmdcolin)Added a --bgzip_fasta option for prepare-refseqs.pl and also the ability to index reference sequence names if they are manually specified as a fasta index e.g.
refSeqs=genome.fai. Thanks to @FredericBGA for the report! (issue #1281, issue #1282, @cmdcolin).
Bug fixes
Fixed issue where some generate-names setups would fail to index features. Thanks to @BioInfoSuite for reporting (issue #1275, issue #1283, @cmdcolin)
Fixed issue with getting feature density from BAM files via the index stats estimation (issue #1233, @cmdcolin)
Fixed issue where some feature mouseovers where not working properly (issue #1236, @cmdcolin)
Fixed issue where instantiating JBrowse via
standalone.jsdidn't work when in a production build with JBROWSE_PUBLIC_PATH overridden (issue #1239, @garrettjstevens)Small fix for issue where SNPCoverage would crash on some feature filters (issue #1241, @cmdcolin)
Fixed issue where JBrowse Desktop was not able to access remote files (issue #1234, issue #1245, @cmdcolin)
Fix issue where the Hierarchical track selector contained a bunch of blank whitespace. Thanks to @nathanhaigh for reporting! (issue #1240, issue #1253, @cmdcolin)
Fixed issue where whitespace surrounding GFF3 attributes and attribute names was incorporated (issue #1221, issue #1254, @cmdcolin)
Fixed issue with some GFF3Tabix tracks having some inconsistent layout of features (issue #1244, issue #1260, @cmdcolin)
Fixed CRAM store not renaming reference sequences in the same way as other stores (issue #1277, @rbuels, @cmdcolin)
Fixed bug where older browsers e.g. IE11 were not being properly supported via babel (issue #1259, issue #1267, @cmdcolin)
Fixed bug where some files were not being fetched properly when changing refseqs. Thanks to @luke-c-sargent for the report (issue #1252)
Fixed storeTimeout on CRAM files being unused which can result in excessive fetches (issue #1235, @cmdcolin)
Fixed issue where JBrowse would load the wrong area of the refseq on startup resulting in bad layouts and excessive data fetches. Thanks to @hkmoon, @cmdcolin, and @garrettjstevens for debugging (issue #1190, issue #1235, pull #1187)
Fixed issue where CRAM layout and mouseover would be glitchy due to ID collisions on features (issue #1271, @cmdcolin)
Fixed parsing of certain bigBed files that were hanging on track startup (issue #1226, issue #1229, @cmdcolin)
Fixed issue where some parts of a CRAM file would not be displayed in JBrowse due to a CRAM index parsing issue (@cmdcolin)
Fixed an issue where BAM features were not lazily evaluating their tags (@cmdcolin)
Notable changes
- Rendering of features in popups, mouseover tooltips, and feature labels were
made to escape HTML. If you are using literal HTML labels in these places
then set the attribute
unsafePopup,unsafeMouseover, orunsafeHTMLFeatureson your tracks. Thanks to @garrettjstevens (issue #1263, @cmdcolin).