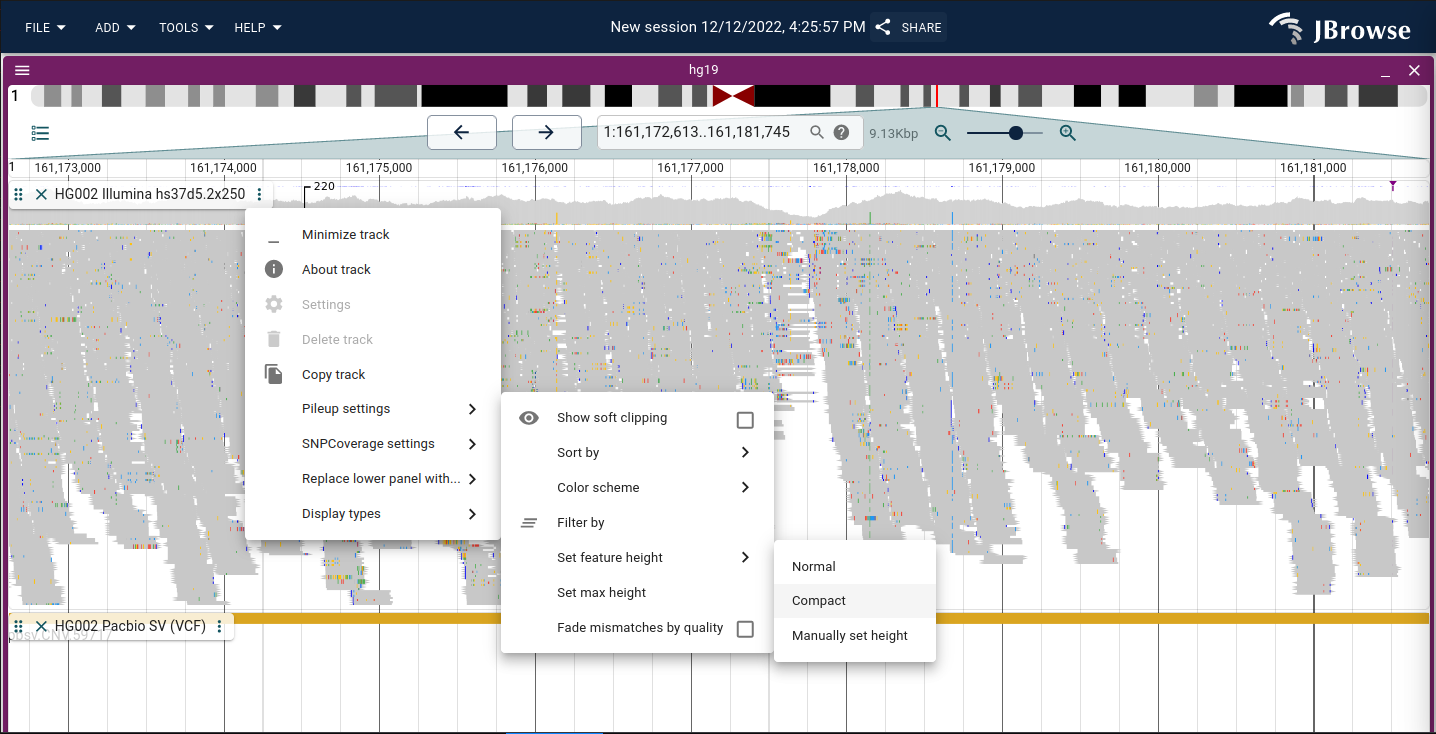
Alignments track
Visualizing alignments is an important aspect of genome browsers. This guide will go over the main features of the "Alignments track." The alignments track is a combination of a pileup and a coverage visualization.
Pileup visualization
The pileup is the lower part of the alignments track and shows each of the reads as boxes positioned on the genome.
By default the reads are colored red if they aligned to the forward strand of the reference genome, or blue if they aligned to the reverse strand.
Coverage visualization
The coverage visualization shows the depth-of-coverage of the reads at each position on the genome, and also draws using colored boxes any occurrence of mismatches between the read and the reference genome, so if 50% of the reads had a T instead of the reference A, half the height of the coverage histogram would contain a 'red' box.
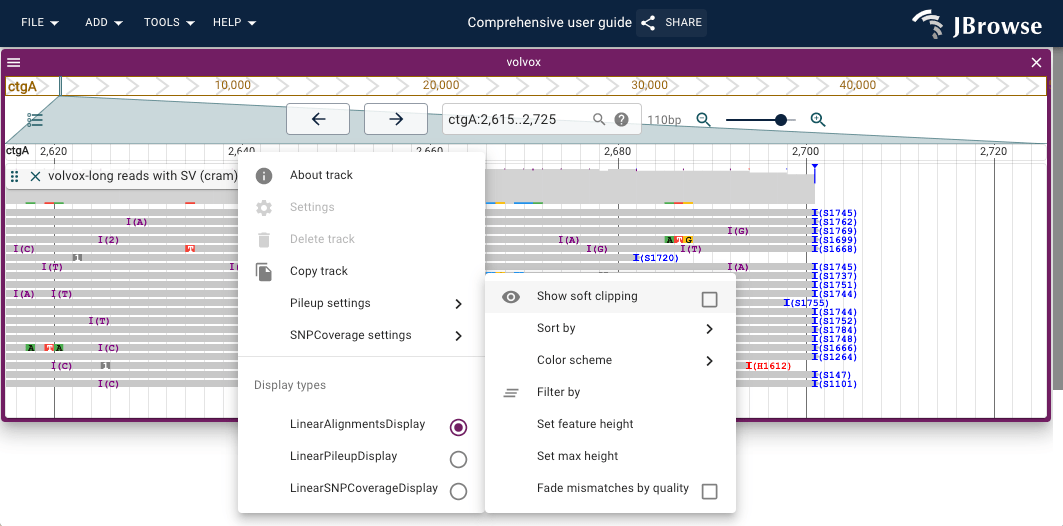
Show soft clipping
If a read contains bases that do not map the the genome properly, they can either be removed from the alignment (hard clipping) or can be included, and not shown by default (soft clipping).
JBrowse 2 also contains an option to "show the soft clipping" that has occurred. This can be valuable to show the signal around a region that contains structural variation or difficult mappability.
Sort by options
The alignments tracks can also be configured to "sort by" a specific attribute for reads that span the center line.
By default the center line is not shown, but by showing it (detailed below) then you will obtain a better idea of what the "sort by" option is doing.
Showing the center line
- Open the hamburger menu in the top left of the linear genome view
- Select "Show center line"
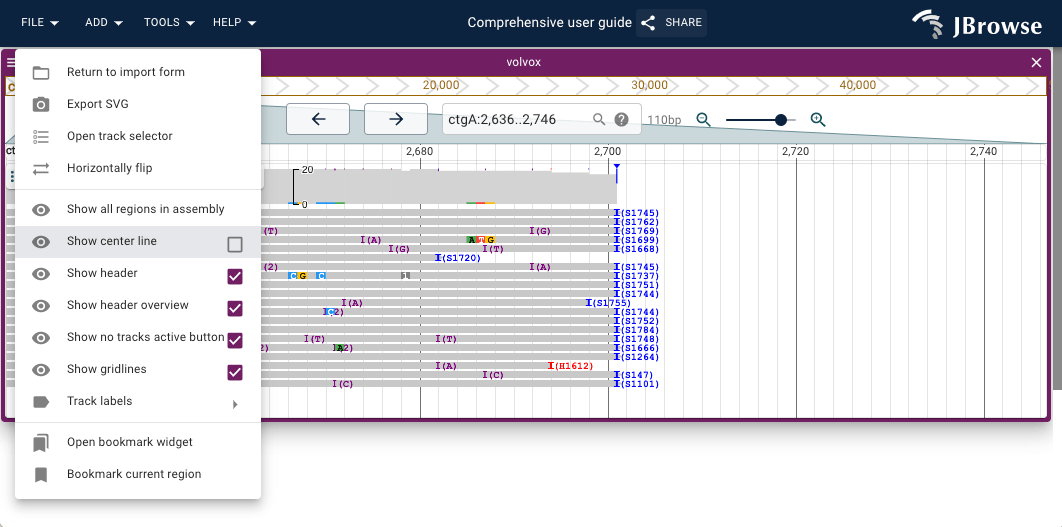
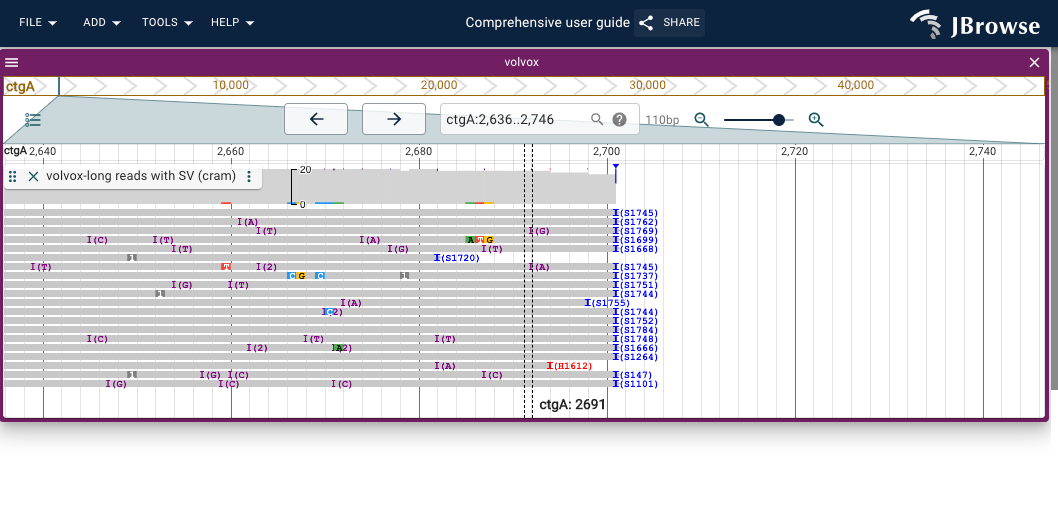
The center line is used by the 'Sort by' function discussed in this section; the sort is performed using properties of the feature, or even exact base pair underlying the center line.
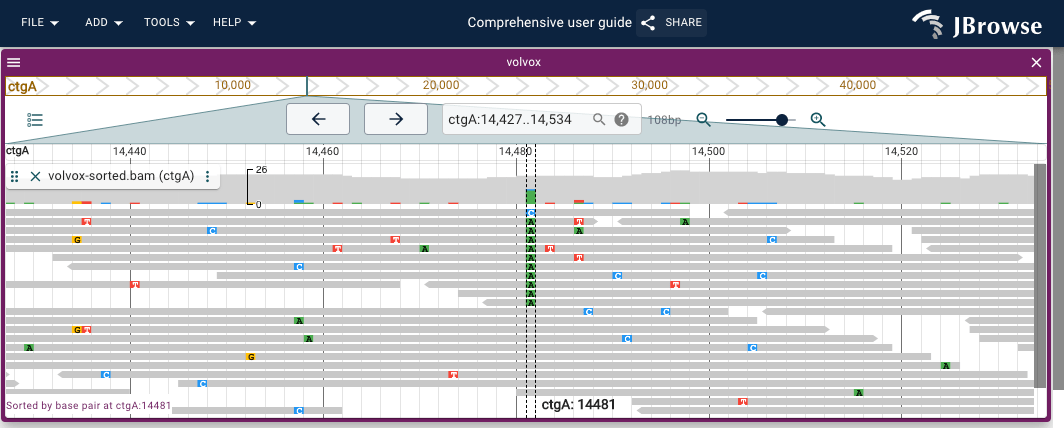
Sorting by base pair
Sorting by base pair will re-arrange the pileup so that the reads that have a specific base pair mutation at the position crossing the center line (which is 1bp wide) will be arranged in a sorted manner. To enable Sort by base pair:
- Open the track menu for the specific track using the vertical '...' in the track label
- Select
Pileup settings->Sort by->Base pair
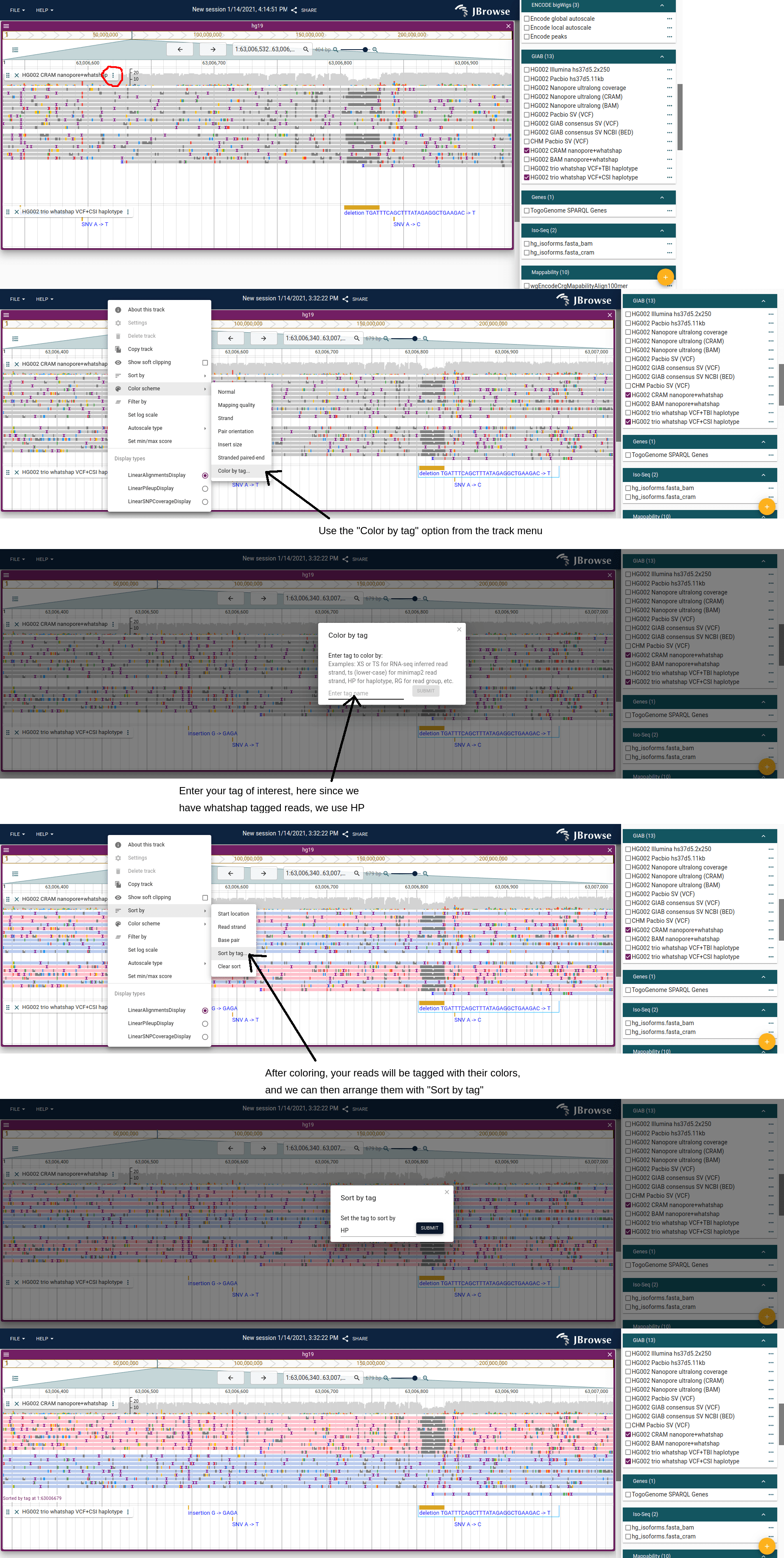
Sort, color and filter by tag
With these features, we can create expressive views of alignments tracks. For example, in the below step-by-step guide, it shows how to color and sort the reads by the HP tag:
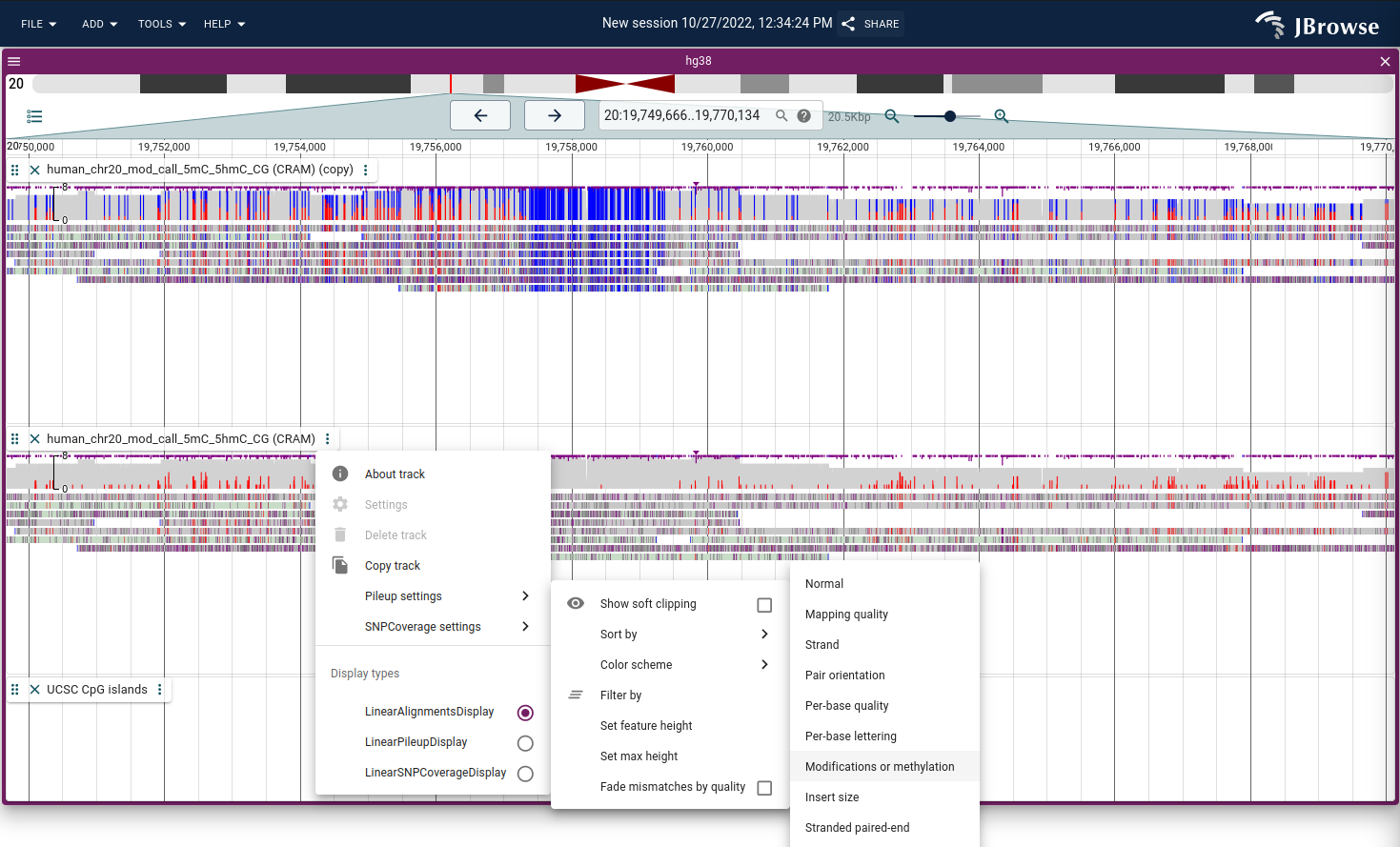
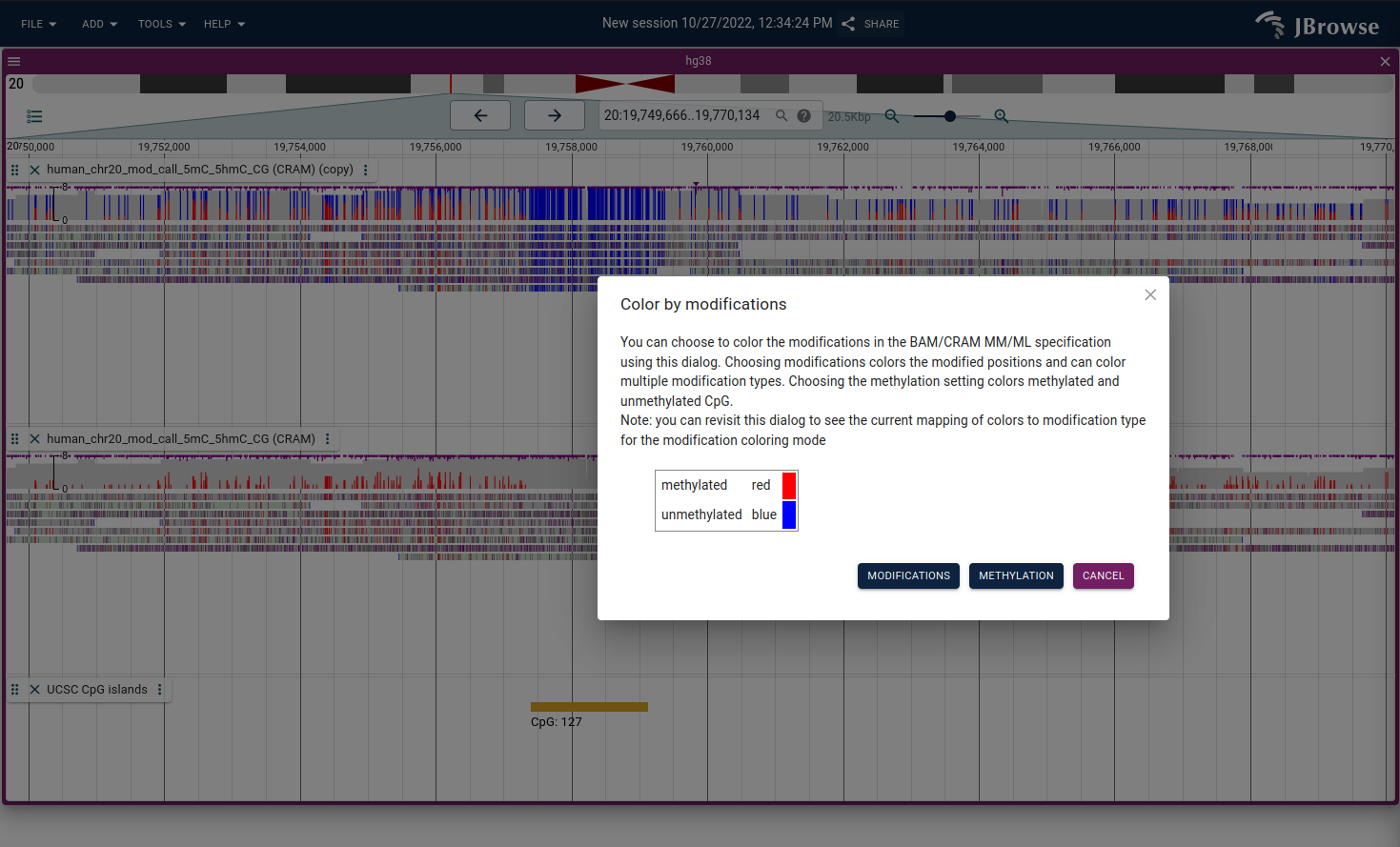
Color by modifications/methylation
If you have data that marks DNA/RNA modifications using the MM tag in BAM/CRAM format, then the alignments track can use these tags to color modifications. It uses two modes:
- All modifications - draws the modifications as they are
- modifications - draws the modifications as they are
- Methylation mode - draws both unmodified and modified CpGs (unmodified positions are not indicated by the MM tag and this mode considers the sequence context)
Color by orientation
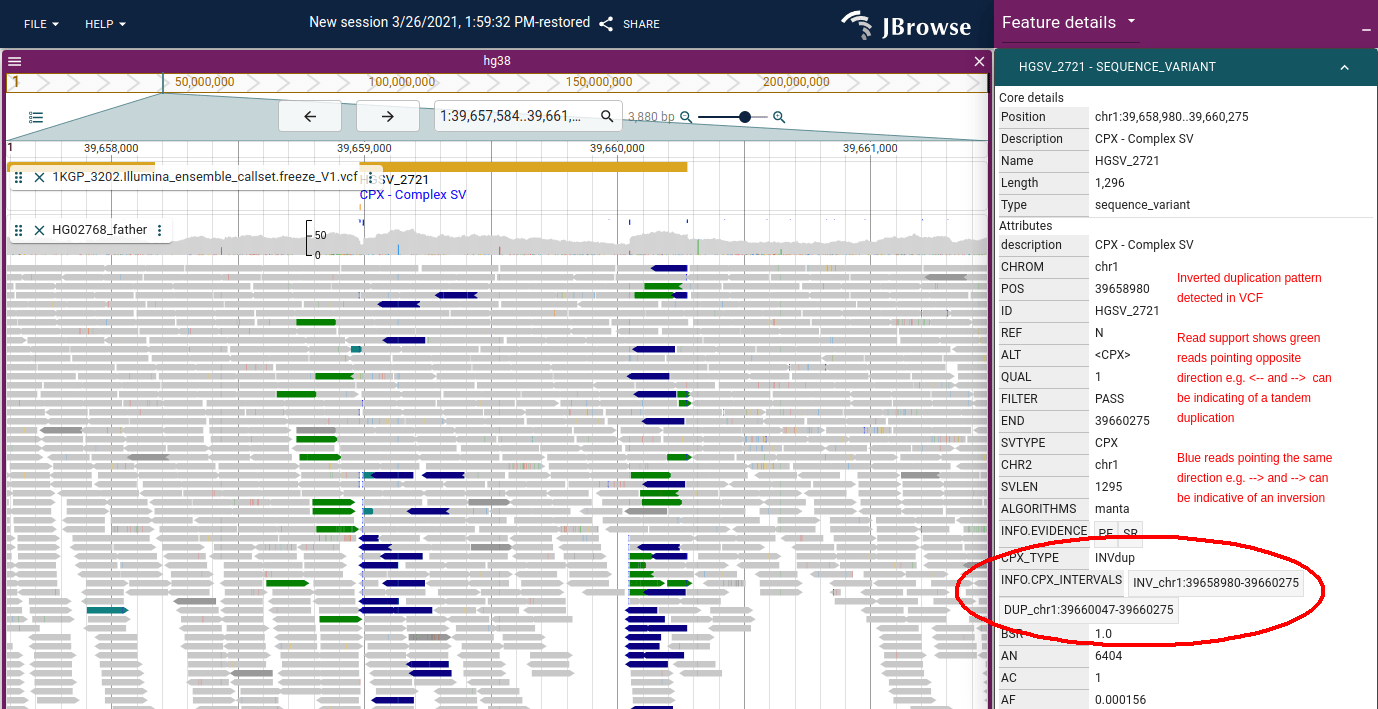
JBrowse uses the same color scheme as IGV for coloring by pair orientation. These pair orientations can be used to reveal complex patterns of structural variation.
See IGV's Interpreting Color by Pair Orientation guide for further details on interpreting these pair orientations.
Sashimi-style arcs
The alignments track will draw sashimi-track style arcs across spliced alignments (indicated by N in the CIGAR string). If the reads additionally are tagged with XS tags, it will try to draw the arcs using the strand indicated by the alignment.
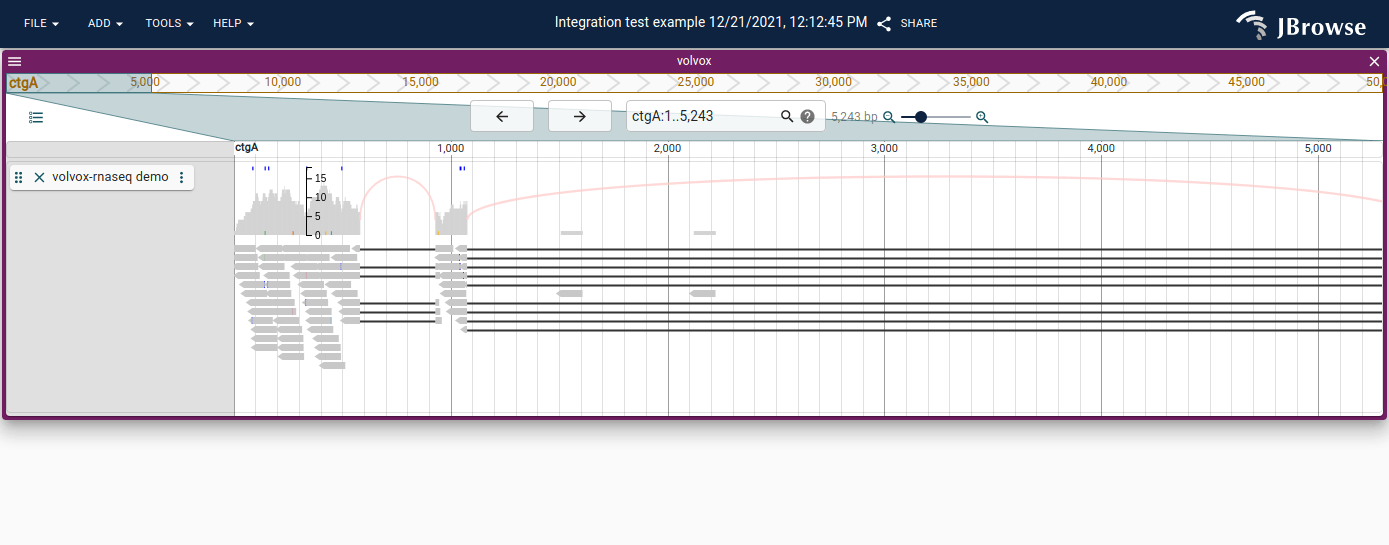
You can disable these by clicking on the track menu (vertical "..." next to track label, then hovering over SNPCoverage options, and unchecking "Draw arcs").
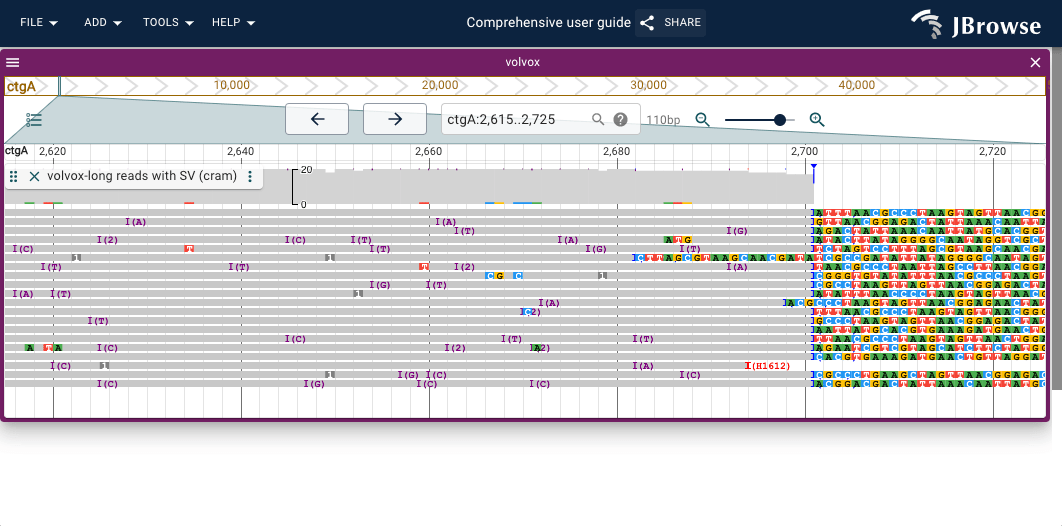
Insertion and clipping indicators
The alignments track will also draw an upside-down histogram of insertion and soft/hard clipped read counts at all positions, and mark significant positions (covering 30% of the reads) with a colored triangle.
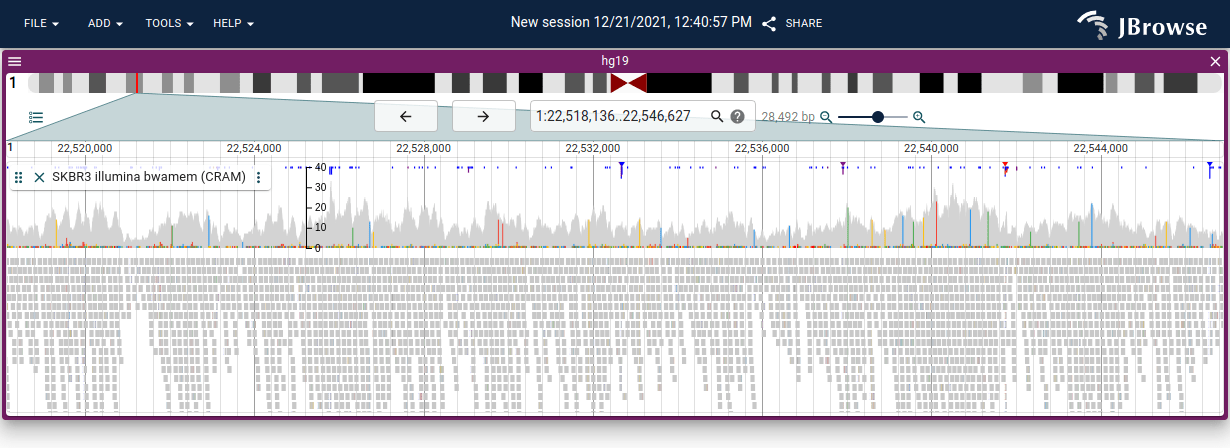
Also, insertions that are larger than 10bp are marked with a larger purple rectangle, seen in the screenshot below. Generally, long reads span larger insertions better, so this feature is more prominent with large reads.
You can disable these by clicking on the track menu (vertical "..." next to track label, then hovering over SNPCoverage options, and unchecking "Draw insertion/clipping indicators" and "Draw insertion/clipping counts").
Using the "Arc display"
In JBrowse 2.3.0, we introduced the ability to render "Arcs" to show long range connections between reads. This information is very valuable for revealing structural variation, misassemblies, etc.
To enable, use the track menu to launch "Display types"->"Arc display" or "Replace lower panel with..."->"Arc display"
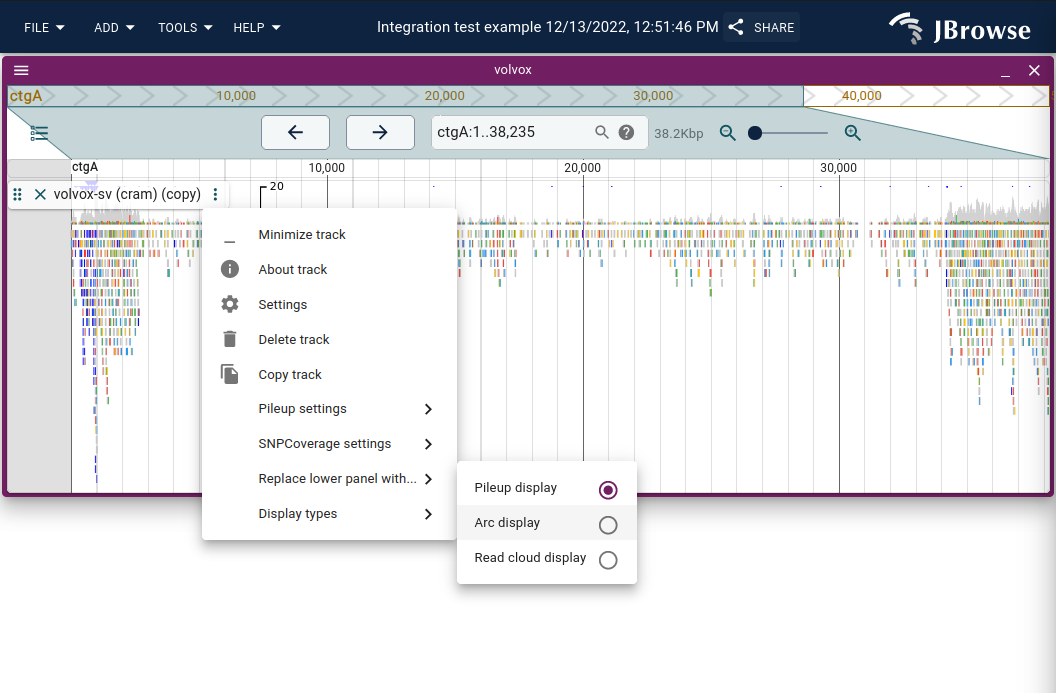
The "Arc display" bezier curves will automatically try to fit into the window when you click and drag the track height, so you can create a dense display of arcs using multiple data tracks
Long range interactions are indicated using vertical lines (connecting to other chromosomes for example) or larger semi-circular arcs for off-screen interactions). You can turn off rendering these events using the track menu if they are not relevant to your interest.
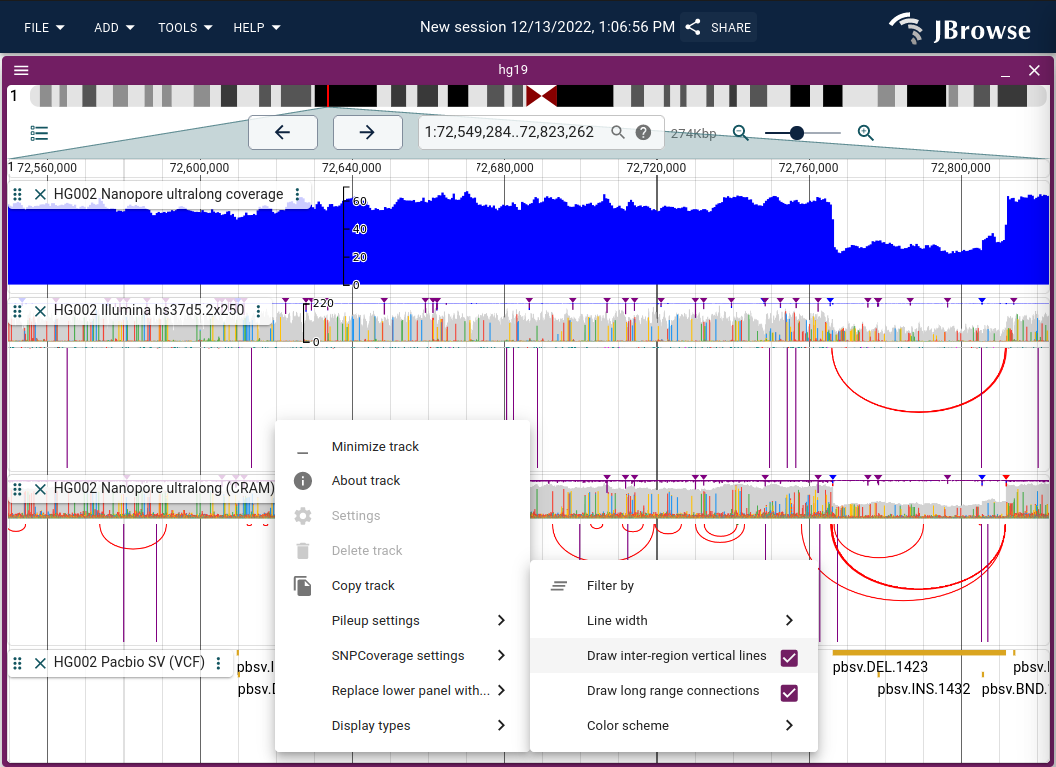
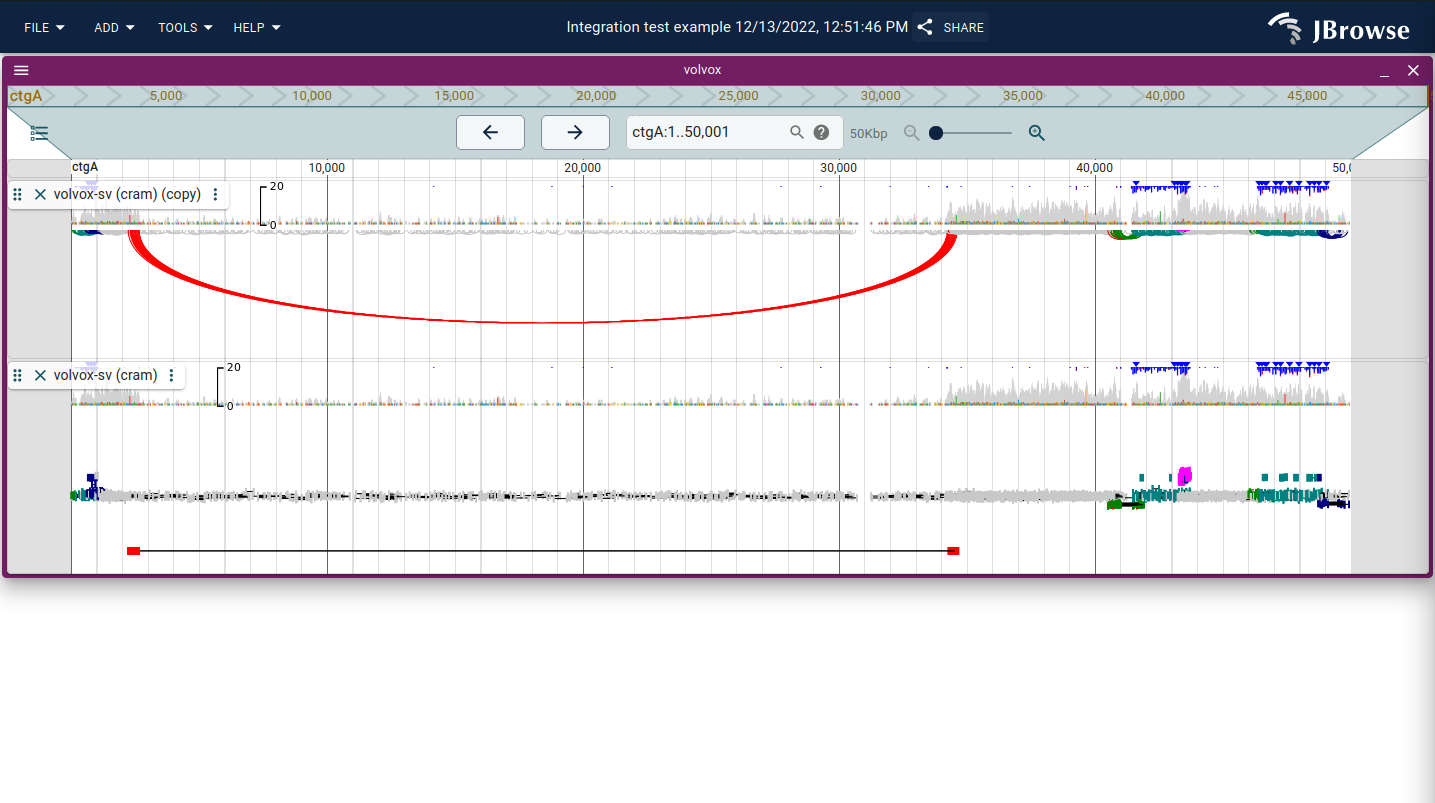
Using the "Linked reads display"
Similar to the "Arc display" we also offer what we call the "Read cloud" display. It is similar in some ways to the "Arc display" but renders paired-end or split read features as connected and stratified by the distance between their connections logarithmically on the "y-position" of the track. Similar to the "Arc display", clicking and dragging the track height of the "Read cloud display" will re-pack features into that area.
Compacting the view of alignments tracks
Users can create a more compact display of alignments using Track menu->Pileup settings->Set feature height->Compact